Acquisition des spectres
Une fois l’échantillon stabilisé à 12% d’humidité, les spectres ont été acquis. Les spectres ont été acquis à l’aide d’un spectromètre proche infrarouge microNIR portatif de 60 g développé par « VIAVI solution » (Photo 6). Sa gamme spectrale est de 950 à 1650 nm (soit 6060 cm-1 à 10526 cm-1 en nombres d’onde), avec une résolution spectrale d’enregistrement de 6,2 nm. Sa température optimale d’utilisation est comprise entre 45 et 55 °C. Les spectres infrarouges ont été obtenus en scannant la face radiale (face RT) des échantillons à une distance fixe de 3mm de l’émetteur de lumière (qui correspond à l’épaisseur du collier attaché à cette dernière). Chaque spectre constitue la moyenne de 100 scans et a été acquis en réflexion diffuse. Selon la dimension des échantillons, deux (2) à quatre (4) spectres ont été pris pour chaque échantillon, et les prises de mesure ont été réparties du centre à la périphérie de la surface considérée afin de considérer la variabilité radiale. Chaque mesure étant distante de 2,5 cm environ (Figure 3). Et pour chaque échantillon, c’est la moyenne de ces 4 mesures qui est considéré, un échantillon étant associé à une unique valeur de référence (infradensité) mesurée au laboratoire.
Traitement des données
Logiciel utilisé Les traitements des données spectrales ont été effectués sur Microsoft Excel et avec le logiciel ChemFlow. Quant à l’analyse de la variabilité, l’ANOVA a été utilisée, avec les logiciels R et XLSTAT.
Préparation des données spectrales Les spectres acquis par l’appareil ont été entre 908 à 1676 nm (5965 cm-1 à 11012 cm-1), mais pour le spectromètre microNIR, c’est la région du 950 à 1650 nm (soit 6060 à 10526 cm-1 en nombre d’ondes) qui est généralement utilisée puisque la zone se trouvant à l’extérieur est généralement bruitée (Razafimahatratra, 2017). Ainsi, les longueurs d’ondes ont été réduites à 112 variables (longueurs d’onde, point de mesure). Après réduction des longueurs d’onde, la moyenne des spectres acquis pour chaque échantillon a été calculée pour ainsi avoir un spectre par échantillon pour le traitement. Enfin, la collection spectrale a été divisée en deux lots : étalonnage et validation. Le jeu d’étalonnage est constitué de deux tiers (2/3) de tous les échantillons (soit 158 parmi les 237 échantillons) et sur lesquels les modèles prédictifs ont été établis. Le jeu de validation est constitué par le reste des données d’apprentissage qui permettra de faire une validation indépendante du modèle prédictif (soit 79 échantillons pour notre étude). Cette division des échantillons en deux lots a été effectuée de manière à inclure les deux valeurs de référence extrême (minimum et maximum) dans le lot d’étalonnage pour plus de variabilité. Pour la division en 2 lots, les échantillons ont été triés dans l’ordre croissant suivant la valeur de référence considérée (valeur de l’infradensité). Pour le lot de validation, tous les un sur trois échantillons à partir du second échantillon, en évitant le dernier, ont été retenu.
Prétraitements Avant l’établissement du modèle, il est recommandé de prétraiter les spectres acquis. Les prétraitements sont des procédés qui permettent de réduire les défauts sur les collections de spectres afin d’améliorer la qualité du signal (Rinnan, Berg & Engelsen, 2009). « En effet, les conditions idéales de la spectroscopie proche infrarouge correspondent à une mesure d’une solution transparente et peu concentrée. Ces conditions se rencontrent au laboratoire, avec des échantillons préparés. Dans ces conditions, la concentration d’un composé d’intérêt est directement reliée à son absorbance. Tous les photons traversent l’échantillon en ligne droite, à l’exception de ceux qui seront absorbés par l’échantillon. La loi de Beer-Lambert stipule que l’absorbance d’un composé à la longueur d’onde λ, notée A(λ), est proportionnelle à la concentration C du composé, à la longueur du trajet optique L, et au coefficient d’extinction du composé à cette même longueur d’onde qui est noté ε(λ) : ?(?) = ?(?)?? Mais en pratique, la SPIR n’est jamais utilisée dans les conditions idéales de la Loi de Beer Lambert. C’est justement son intérêt de pouvoir être utilisée sur des produits bruts, tels que de la farine, du bois, des sols, etc. La relation entre le spectre et la concentration est alors plus compliquée. La loi de Beer-Lambert n’est plus vérifiée. Dans le cas d’un échantillon réel, le spectre est perturbé par la diffusion des photons et par le bruit de mesure. La diffusion des photons : (1) allonge la longueur moyenne du trajet optique d’un facteur k ; (2) conduit un certain nombre de photons à échapper au capteur, ce qui ajoute un terme de fuite : A f(λ). Le bruit de mesure est dû à un ensemble de phénomènes aléatoires présents tout au long de la chaîne de mesure (lumière parasite, bruit électronique du capteur, etc.). Il se traduit par un terme additif A b(λ). Finalement, l’absorbance réellement mesurée est affectée d’un terme multiplicatif et de 2 termes additifs. L’équation de l’absorbance devient :
A(λ) = kε(λ)LC + A f(λ) + A b(λ)
Les différentes stratégies des prétraitements vont alors consister à corriger au moins l’un des trois termes : k, A f(λ), A b(λ) et tentent de se rapprocher de la seule contribution du paramètre recherché ». (Roger & Ecarnot, 2016) Il existe différents types de prétraitements, mais dans cette étude a principalement opté pour :
– le « de-trending » qui permet de corriger, de réduire la ligne de base des spectres, en enlevant la tendance d’augmentation d’absorbance lorsque les nombres d’onde augmentent, qui est un phénomène naturel dans la région spectrale du proche infrarouge (Barnes, Dahona & Lister, 1989). Cette méthode consiste à modéliser la tendance globale du spectre par un polynôme (de différents degrés) et de soustraire au spectre ce polynôme pour corriger cette tendance croissante. Dans cette étude, ce sont des polynômes d’ordre 2 qui ont été utilisés.
– la « Standard Normal Variate » qui consiste à enlever les erreurs et bruits dus à l’effet additif et aussi à l’effet multiplicatif (Barnes, Dahona & Lister, 1989). Il s’agit de centrer et réduire les spectres.
– les dérivées avec le lissage des spectres : il s’agit dans l’étude de la méthode de lissage et de dérivation de Savitsky-Golay. D’abord, la méthode identifie un polynôme (de plusieurs degrés) sur une fenêtre d’une certaine largeur (W) et centré sur le point à calculer. Ensuite, le polynôme est dérivé analytiquement et c’est la valeur de sa dérivée qui est utilisée (Savitzky & Golay, 1964). L’algorithme de Savitziky-Golay de premier ordre et de second ordre et en variant la largeur de la fenêtre W a été appliqué. Celui qui donne un meilleur résultat a été ensuite retenu. Des simulations pour identifier la meilleure largeur de la fenêtre de mesure ont donc été effectuées. Pour la dérivée première (SG1), la largeur de la fenêtre glissante est de 7 points et en utilisant un polynôme d’ordre 2, de même pour la dérivée seconde (SG2), la largeur de la fenêtre est de 7 points mais en utilisant un polynôme d’ordre 3. (Annexe 6). Les dérivées avec le lissage des spectres de Savitsky-Golay enlèvent généralement les effets multiplicatifs. Avant l’établissement des modèles, ces différents types de prétraitements ont été appliqués aux spectres, isolément, puis combinés 2 à 2 : SNV+Detrend, Detrend+SG1, Detrend+SG2, SNV+SG1, SNV+SG2 et enfin combinés 3 à 3 : SNV+Detrend+SG1 et SNV+Detrend+SG2. Les modèles établis en utilisant les spectres ayant subi ces prétraitements ont été comparés entre eux, puis avec le modèle établi avec les spectres sans prétraitement. Et c’est le meilleur modèle, et donc associé au meilleur prétraitement qui a été retenu.
Sélection de variables : sélection de longueurs d’onde
Toutes les longueurs d’onde dans le proche infrarouge ne sont pas toutes hautement informatives vis-à-vis de la propriété à prédire ; de plus, plusieurs longueurs d’onde sont corrélées. Il peut être intéressant de n’utiliser qu’un nombre réduit de longueurs d’onde, celles les plus informatives pour avoir des modèles de prédiction de bonne qualité. De plus, cela améliore la robustesse du modèle. La méthode de sélections de variables cherche donc à savoir quelles sont les longueurs d’onde qui jouent un rôle important dans le phénomène étudié (Gauchi & Chagnon, 2001). Des sélections de longueurs d’onde ont été effectuées sur les spectres prétraités. Elle a été réalisée grâce à la fonction CovSel du logiciel Chemflow, qui sélectionne les longueurs d’ondes associées aux covariances les plus élevées (covariance entre les absorbances de la longueur d’onde avec les valeurs de référence mesurées). La méthode utilisée comporte 6 étapes (itérations) de façon à éliminer en moyenne 20 longueurs d’onde à chaque itération. D’abord, un modèle est établi sur la base de toutes les longueurs d’onde. Dans la deuxième étape, un modèle est établi sur la base de 100 longueurs d’onde associées aux covariances les plus élevées dans la première étape, puis le modèle ne considère que 80 variables, 60 variables, 40 variables et 20 variables parmi les longueurs d’ondes. Enfin, les modèles basés sur 112 variables, 100 variables, 60 variables, 40 variables et 20 variables sélectionnées sur le spectre non prétraité et sur chacun des spectres prétraités ont été comparés entre eux. Et ce sont les x variables (longueurs d’onde) correspondant au meilleur modèle qui sont donc considérés.
. Identification des prétraitements adéquats pour la modélisation
Pour chaque prétraitement, le meilleur modèle associé au meilleur nombre de longueurs d’onde est déjà identifié (tableau 6). Parmi ces modèles, le meilleur modèle est celui obtenu avec les prétraitements « SNV+DT ». Il est associé au minimum de RMSEcv, qui est égal à 0,026, à la valeur de R² en validation croisée la plus élevée, qui est égale à 0,69, et au maximum de RPD égal à 1,74 (Tableau 6; Figures 11, 12, 13). Par contre, les modèles les moins performants, avec le maximum de RMSEcv, qui est de 0,029, sont obtenus avec les spectres non prétraités et avec les prétraitements DT+SG1. Ils ont donnés des valeurs de R² en validation croisée comprises entre 0,51 et 0,54, et un RPD égal à 1,56 (Tableau 6; Figures 11, 12, 13). Ainsi, le choix du meilleur prétraitement pour le modèle de l’infradensité basé sur les spectres du microNIR se porte sur la combinaison de « SNV+ DT » (Tableau 6; Figures 11, 12 et Figure 13).
La sélection de longueurs d’onde
Plusieurs études ont démontré que la sélection de longueurs d’onde améliore considérablement la qualité du modèle. A l’exemple des études de Diesel et al. en 2014 sur l’estimation de la densité de Mimosa tenuiflora par la méthode SPIR et qui a démontré que le modèle considérant seulement 17 longueurs d’onde (de 2 090 à 2 208 nm) a donné une bonne prédiction par rapport au modèle qui utilise toutes les longueurs d’onde. Il y a aussi l’étude de Razafimahatratra en 2017 qui a établi que les modèles avec sélection de longueurs d’onde ont été significativement meilleurs que les modèles établis sans sélection, concernant la prédiction de plusieurs propriétés chimiques du bois de plusieurs espèces d’eucalyptus. Le choix du nombre de longueurs d’onde à utiliser doit se baser sur la valeur de la RMSEcv donnée par le modèle associé, et qui doit être la plus faible. Dans cette étude, pour chaque prétraitement, les plus faibles RMSEcv sont associés aux nombres de longueurs d’onde compris entre 60 et les 112 variables. Mais c’est le modèle basé sur les spectres prétraités avec méthode « SNV+Dt » et considérant toutes les longueurs d’onde (donc sans sélection de variables) qui a été retenu par le fait que c’est le modèle qui est associé à la valeur la plus faible de RMSEcv. Pour les autres prétraitements, les modèles avec sélection de longueurs d’onde (au nombre de 60 à 80) utilisés dans cette étude sont associés soit à un nombre de variables latentes (VL) assez faible, soit à un nombre de VL élevées (jusqu’à 15). Pour certains, des nombres de VL faibles ont été retenus car des nombres de VL élevés seraient associés à un mauvais critère de Durbin Watson. Mais lorsque le nombre de variables latentes est inférieur à ce qu’il faut, le modèle peut manquer de variables prédictives nécessaires à l’explication de la variable réponse et peut entraîner un sous-ajustement du modèle (« underfitting ») (Sabatier, Reynes & Vivien, 2016). De même, l’erreur de prédiction en validation augmente quand le modèle comporte plusieurs dimensions (Sabatier, Reynes & Vivien, 2016) puisque le modèle devient trop spécifique aux échantillons d’étalonnage et non à d’autres échantillons (Annexe 9). En effet, elle commence à inclure des erreurs et non des informations utiles du spectre. Et un des inconvénients des modèles avec sélection de variable est que les modèles sont très particuliers aux jeux de données d’étalonnage (espèce) et sont plus difficiles à appliquer pour d’autres échantillons ayant des caractéristiques différentes, d’où la nécessité de faire des essais de transfert de calibration entre espèces si une étude veut utiliser le modèle pour prédire sur d’autres espèces. .
|
Table des matières
1. INTRODUCTION
2. METHODOLOGIE
2. 1. Problématique de la recherche et hypothèses
2.1.1. Problématique
2.1.2. Hypothèses
2.1.3. Objectifs de la recherche
2.2. Matériels et méthodes
2.2.1. Matériels d’étude
2.2.2. Origine des échantillons
2.2.3. Mode de collecte des échantillons
2.2.4. Nombre des échantillons
2.2.5. Méthodes
2.2.5.1. Mesure des valeurs de référence en laboratoire
2.2.5.2. Collecte des spectres
Préparation des échantillons
Acquisition des spectres
2.3. Traitement des données
2.3.1. Logiciel utilisé
2.3.2. Préparation des données spectrales
2.3.3. Prétraitements
2.3.4. Sélection de variables : sélection de longueurs d’onde
2.3.5. Elimination des valeurs aberrantes ou « outliers »
2.3.6. Etablissement du modèle de prédiction
2.3.7. Analyse de la variabilité radiale de l’infradensité
3. RESULTATS
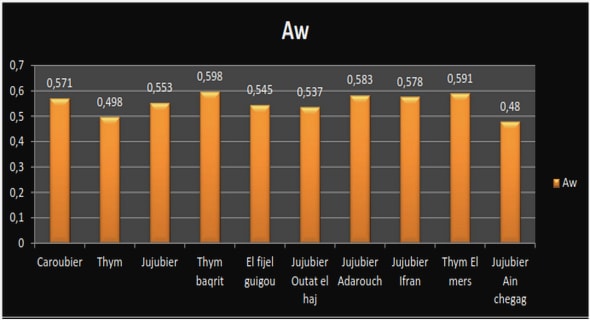
3.1. Analyse exploratoire des valeurs de référence mesurées en laboratoire
3.1.1. Analyse globale de toutes les valeurs de références
3.1.2. Analyse exploratoire des valeurs de référence par sites
3.2. Analyse exploratoire des spectres
3.2.1. Allure générale des spectres
3.2.2. Analyse en composantes principales des spectres bruts
3.3. Sélection de longueurs d’onde
3.4. Identification des prétraitements adéquats pour la modélisation
3.5. Elimination des outliers
3.6. Performance du modèle de prédiction
3.6.1. Etalonnage du modèle et validation croisée
3.6.2. Validation du modèle (validation indépendante)
3.7. Prédiction de l’infradensité : analyse de la varibilité
3.7.1. Analyse de la variabilité intra-arbres de l’infradensité à partir des valeurs prédites
3.7.2. Analyse de la variabilité inter-compartiment de l’infradensite à partir des valeurs prédites
3.7.3. Analyse de la variabilité inter-provenance et inter-variété de l’infradensité à partir des valeurs prédites
4. DISCUSSIONS
4.1. Les échantillons de référence
4.2. Les échantillons d’étalonnage et de validation
4.3. La sélection de longueurs d’onde
4.4. Les prétraitements sélectionnés
4.5. La performance du modèle
4.6. Comparaison avec les études effectuées sur des produits ligneux
4.7. La variabilité de l’infradensité radiale
4.8. Vérification des hypothèses
5. RECOMMANDATIONS
5.1. Sur l’amélioration du modèle
5.2. Sur l’utilisation de Ravenala madagascariensis
6. CONCLUSION
REFERENCES BIBLIOGRAPHIQUES
ANNEXES
![]() Télécharger le rapport complet
Télécharger le rapport complet