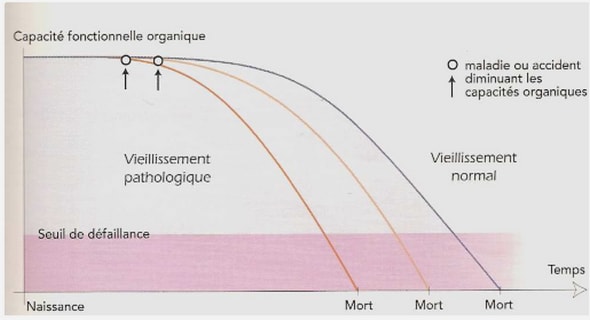
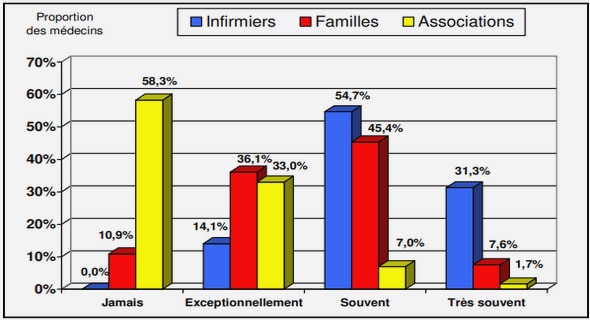
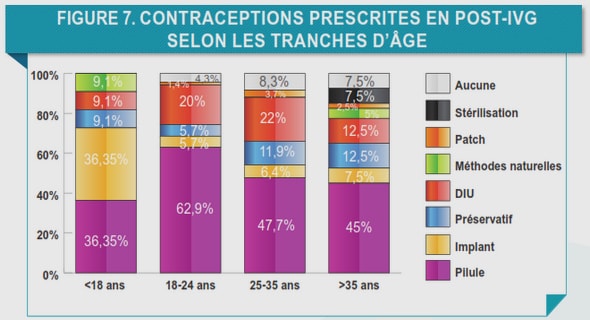
Télécharger le fichier pdf d’un mémoire de fin d’études
Classification des crises épileptiques
Epilepsies avec signes moteurs
Concerne la musculature sur toutes ses formes.
L’événement moteur pourrait consister en une hausse (positive) ou baisse (négative) dans la contraction musculaire pour produire un mouvement.
Les signes moteurs élémentaires
Un type unique de contraction d’un muscle ou d’un groupe de muscles qui est d’habitude stéréotypé et non décomposable en phases.
o Tonique
Une augmentation soutenue dans la contraction musculaire qui dure quelques secondes voire quelques minutes
• Spasme épileptique (jadis spasme infantile)
• Postural
• Versif
• Dystonique
o Myoclonique
Contraction d’un muscle ou d’un groupe de muscles d’une topographie variable (axial, proximal, distal) soudaine et brève (100ms) involontaire, unique ou multiple.
• Myoclonique négative
• Clonique
• Marche jacksonienne
• Tonico-clonique
Une séquence consistant en une phase tonique suivie d’une phase clonique. Des variantes telles que clonico-toniques peuvent être vues.
o clonico-tonique généralisée
Jadis appelée crise grand mal : contraction clonique bilatérale des muscles somatiques, d’habitude associée aux phénomènes autonomiques.
o Atonique
Une perte soudaine ou diminution du tonus musculaire sans myoclonies précédentes apparentes ou un événement tonique durant une à deux secondes, impliquant la tête, le tronc, la mâchoire ou un membre de la musculature.
o Astatique
Une perte de posture érectile, qui résulte d’un mécanisme tonique myoclonique, atonique.
o Synchrone (asynchrone)
Un événement moteur ayant lieu en même temps ou pas dans les ensembles corporels.
Automatisme
C’est une activité motrice plus ou moins coordonnée répétitive, qui a souvent lieu lorsque la cognition est altérée et pour laquelle le sujet est d’habitude amnésique après. Cela ressemble parfois à un mouvement volontaire et peut consister en une continuation inappropriée de l’activité motrice pré-ictus courante.
o Oro-alimentaire
Un léchage ou pincement des lèvres, grincement des dents ou avalement.
o Mimétique
Une expression faciale suggérant un état émotionnel, parfois la frayeur.
o Manuel ou pédestre
– Indique principalement des composantes distales, bilatérales ou unilatérales.
– Tâtonnement, perçage, mouvement de manipulation.
o Gestuel
Parfois unilatéral :
– Tâtonnement ou mouvements exploratoires avec main dirigée soit vers soi soit vers son entourage ou environnement.
– Mouvements ressemblant à ceux destinés à prêter un ton émotionnel supplémentaire au discours.
o Hypercinetique
– Concerne principalement les membres proximaux ou les muscles axiaux qui produisent des mouvements ballistiques, séquentiels et irréguliers.
– Augmentation du taux de mouvement en exécution ou une exécution rapide et inappropriée d’un mouvement.
o Hypocinétique
Une baisse d’amplitude ou du taux ou l’arrêt de l’activité motrice en cours.
o Dysphasique
Une communication langagière altérée sans dysfonctionnement moteur primaire concerné ou des voies sensorielles, qui se manifeste par des troubles de la compréhension, l’anosmie, des erreurs paraphrasiques ou une combinaison de ces phénomènes.
o Dyspraxique
Une incapacité à exécuter spontanément les mouvements appris ou commandés ou limités malgré la non altération des systèmes sensoriels et moteurs concernés, malgré une coopération et une compréhension adéquates
o Gélastique
Un éclat de rire ou fou rire, d’habitude sous un ton affectif approprié.
o Dacrystique
Des éclats de cris de type
– vocal,
– verbal ou
– spontané.
EPILEPSIES AVEC SIGNES NON MOTEURS
L’aura
C’est un phénomène subjectif de l’ictus, qui pour un patient donné peut précéder une crise observable ; lorsqu’elle est isolée, elle constitue une crise sensorielle.
L’aura Sensoriel
Une expérience perceptuelle qui n’est pas causée par un stimulus approprié de l’environnement, modifie la crise de l’aura. Elle peut être :
• élémentaire
Un phénomène informe et unique impliquant une modalité sensorielle primaire.
Elle est :
o somatosensorielle o visuelle
o auditive o olfactive o gustative
o épigastrique o céphalique o automatique.
Elle peut être:
• expérimentale
Un phénomène perpétuel, mnémonique ou affectif incluant des événements hallucinatoires composites ou illusoires. Ces phénomènes peuvent être isolés ou combinés. En outre, il y a le sentiment d’être étranger à soi même. Ces phénomènes ont des qualités subjectives. Ils sont similaires à ceux
expérimentés dans la vie, mais sont reconnus par le sujet comme se passant dans
un contexte hors de la réalité.
Ils peuvent être d’ordre :
o affectif
o mnémonique o hallucinatoire o illusionnel
Dyscognitif
Ce terme décrit des événements dans lesquels le dérangement de la cognition est la caractéristique qui domine ou celle qui est la plus visible, et deux ou plusieurs de ces composantes sont concernées ; l’implication de telles composantes reste indéterminée. Autrement, utiliser le terme le plus spécifique (crise expérimentale mnémonique ou crise expérimentale hallucinatoire). Les composantes de la cognition sont :
o perception : conception symbolique de l’information sensorielle,
o attention : sélection appropriée d’une perception ou tâche principale,
o émotion : signification affective appropriée d’une perception
o mémoire : capacité à emmagasiner et à restituer les
perceptions et les concepts,
o fonction exécutive : anticipation, sélection, surveillance des conséquences et initiation d’activités incluant la praxie, les discours.
L’EVENEMENT AUTONOMIQUE
L’aura autonomique
La sensation compatible avec l’implication du système nerveux autonome, incluant les fonctions cardiovasculaires, gastro-intestinales, sudoromotrices, vasomotrices et thermorégulatrices.
La Crise autonomique
Une altération distincte et objectivement documentée de la fonction du système nerveux autonome incluant les fonctions cardiovasculaires, pupillaires, gastro- intestinales, respiratoires et thermorégulatrices.
LES SIGNES MODIFICATEURS SOMATOTOPIOUES
La latéralité
¾ Unilatérale
C’est une implication exclusive ou virtuelle d’un seul coté en tant que phénomène moteur, sensoriel ou autonomique.
¾ Hémicorporel
D’un coté, droite ou gauche
¾ Généralisé
Plus du minimum de l’implication de chaque coté en tant que phénomène autonomique, sensoriel ou élémentaire.
Composantes motrices modifiées d’avantage comme :
o Asymétriques o Symétriques
La Partie du corps
Cette partie fait référence aux parties concernées (par exemple bras, jambes, visage, tronc et autres).
La Centralité
Modificatrice décrite en proximité à l’axe du corps.
Elle est axiale
Concerne le tronc y compris le cou.
o partie proximale du membre
Signifie l’implication des épaules aux poignets, des hanches aux chevilles.
o partie distale du membre
Indique l’implication des doigts, des mains, des oreilles, des orteils et/ou des pieds.
L’EPIDEMIOLOGIE
Dans le monde, la maladie épileptique a une répartition géographique variable, avec un taux de prévalence de 1,5 à 5%0 habitants. Cette répartition dépend du niveau de développement socio-économique. La prévalence est 5 à 10fois plus élevée dans les pays en voie de développement que dans les pays industrialisés. Le syndrome de West est l’encéphalopathie épileptique le plus fréquent.
Dans le monde, la prévalence du syndrome de West est d’environ 1/4000 à 1/6000. Statistiquement les garçons sont plus à risques que les filles, dans un ratio de 3/2. Dans 90% des cas les spasmes se manifestent pour la première fois entre 3 et 12 mois de vie. Dans des cas plus rares, les spasmes débutent pendant les deux premiers mois de vie ou durant la deuxième à la quatrième année. (J Roger et al, 2002)
En Afrique, la maladie épileptique a une répartition géographique variable, avec un taux de prévalence de 14%0 en moyenne. A Madagascar la fréquence varie de 12 %0. En milieu intertropical, il y a une grande variabilité avec des zones de moyenne prévalence (8 à12 %0 pour le Sénégal, la Cote d’Ivoire et le Togo) et d’autres de forte prévalence (20%0 voire plus de 40%0 pour le Liberia, le Nigeria, et la Tanzanie). La prévalence est relativement faible au nord et au sud du continent africain. Elle varie de 4 à 8%0. (I. Mbodj, 2003)
Une étude camerounaise sur le syndrome de West a montré que la prédominance était masculine (59,46%), avec un sexe ratio de 1,47. un facteur étiologique était retrouvé dans 83,78% des cas.
LES FACTEURS ETIOLOGIQUES
Une lésion cérébrale préexistante peut être mise en évidence dans 60-90 pour cent des cas (Diebler et Dulac, 1987 ; Ludwig, 1987). L’histoire peut révéler une lésion acquise pré-, péri- ou postnatale, et l’examen clinique les anomalies d’un syndrome neurocutané, mais au moment du diagnostic, l’examen neurologique proprement dit est habituellement peu contributif. Par conséquent, la neuroradiologie est une source majeure d’informations, mais elle doit être réalisée avant le début de la corticothérapie qui donne des images d’atrophie cérébrale (Gastaut et al., 1978) difficiles à distinguer d’une atrophie qui est elle-même présente dans la moitié des cas (Curatolo et al., 1982 ; Singer et al., 1982) et des variations physiologiques des espaces péricérébraux chez le nourrisson (Fukuyama et al., 1979).
L’imagerie par résonance magnétique (IRM) peut en outre méconnaître des anomalies de la substance grise au cours du second semestre de la vie, et devrait par conséquent être réalisée après l’âge de 18 mois (Valk et Knaap, 1989). La signification d’une image de retard de myélinisation est difficile à déterminer en l’absence de données neuropathologiques (Muroi et al., 1996). Les investigations neuropathologiques post-mortem ont été négatives dans seulement 3-4 pour cent des cas, mais il y a un biais évident en faveur des cas les plus sévères.
Les malformations cérébrales concernent 30 pour cent des cas si on inclut les cas de syndrome neurocutané. Les plus fréquentes sont l’agénésie du corps calleux, y compris le syndrome d’Aircardi (Diebler et Dulac, 1987), la polymicrogyrie (Gastaut et al., 1978), la lissencéphalie (Bordarier et al., 1986), l’hémimégalencéphalie (Robain et al., 1989), la dysplasie corticale focale (Chugani et al., 1990) et la schizencéphalie. Il reste à préciser si la découverte de microdysgénésie a une réelle signification pathogène (Jellinger, 1987 ; Lyon et Gastaut, 1985). Les malformations vasculaires, y compris la maladie de Sturge-Weber (Millichap et al., 1962) et les fœtopathies (Watanabe et al., 1973) sont des causes rares. L’évidence précise de l’ischémie périnatale responsable de SW est vraisemblablement plus proche de 15 pour cent (Aircadi et Chevrie, 1978) que des 80 pour cent antérieurement annoncés, et les lésions affectent le cortex, la substance blanche et les noyaux gris centraux plus souvent que le tronc cérébral (Van Bogaert, 1993). L’hypoglycémie peut être la cause en elle-même du SW ou parce qu’elle est la conséquence d’une hypotrophie fœtale causée par une hémorragie utérine ou une toxémie gravidique (Riikonen et Donner, 1979).
Les lésions post-natales causales incluent l’ischémie, l’infection et les traumatismes (Millichap et al., 1962 ; Ganji et al., 1987 ; Aubourg et al., 1985). Les erreurs innées du métabolisme sont rarement en cause, mais une grande diversité d’entre elles peut produire des convulsions infantiles, parmi lesquelles les spasmes sont simplement d’un type particulier.
Néanmoins, la maladie de Menkes (Sztriha et al., 1994), la phénylcétonurie et certaines mitochondriopathies (Makela-Bengs et al., 1995) produisent un taux élevé de spasmes infantiles. En ce qui concerne la dernière étiologie, elle est facilement reconnue au vu de l’hypersignal en T2 qui dessine les noyaux gris centraux. Tous les types de tumeurs du nourrisson, y compris les papillomes du plexus choroïde associés au syndrome d’Aicardi, peuvent s’accompagner des spasmes infantiles.
Une encéphalite herpétique néonatale ou du nourrisson constitue un risque élevé de SW. L’incidence du SW après méningite bactérienne est plus basse. La responsabilité de la vaccination contre la coqueluche a pu être exclue à la suite d’une analyse épidémiologique portant sur les conséquences d’un changement du calendrier des vaccinations au Danemark (Bellman et al., 1983).
Sept à 17 pour cent des patients ont une histoire familiale d’épilepsie ou de convulsions fébriles (Jan et al., 1971), l’incidence étant de 40 pour cent dans les formes cryptogéniques (Matsumoto et al., 1981). Il y a une transmission autosomique dominante dans la sclérose tubéreuse de Bourneville (Wilson et Carter, 1978), la neurofribromatose (Motte et al., 1993) et le syndrome de CHARGE (Curatolo et al., 1983).
La transmission est dominante liée au sexe dans l’incontinentia pigmenti et le syndrome d’hétérotopie en bande/lissencéphalien (des Portes et al., 1998). D’autres familles ont une forme liée au chromosome Xp11.4-Xter (Claes et al., 1997) ou Xp11.4Xp22.11 (Stromme et al., 1999). Une translocation chromosomique concerne le syndrome de Williams (Mizugishi et al., 1998) et la trisomie 21 (Silva et al., 1996). Dans cette dernière, le SW tend à résister au traitement quand celui-ci est instauré tardivement.
L’association d’une dysmorphie, d’une hypotonie et des signes pyramidaux est observée en cas d’intervention de la duplication du chromosome 15q (Bingham et al., 1996). Diverses dysmorphies familiales sont associées aux spasmes infantiles, y compris les pouces larges (Tsao et al., 1990), et le bec de lièvre avec exophtalmie (Tutuncuoglu et al., 1996).
Il a été rapporté des spasmes infantiles familiaux avec microcéphalie et syndrome néphrotique (Roos et al., 1987). Une augmentation modérée de l’incidence du SW a été observée dans les familles des enfants avec SW cryptogénique mais le risque estimé de récurrence chez un frère ou une sœur est inférieur à 1 pour cent (Dulac et al., 1993).
Dans quelques familles finlandaises, une hypsarythmie était associée à une encéphalopathie congénitale, avec œdème et atrophie optique (PEHO), transmise sur un mode autosomique récessif (Salonen et al., 1987).
LES ASPECTS ELECTROCLINIQUES
L’hétérogénéité de la présentation clinique de l’EEG, de l’étiologie et de l’évolution appellent un démembrement du groupe à la recherche de corrélation entre une cause particulière, sa présentation clinique et les tracés EEG correspondants. Une telle corrélation existe pour certaines malformations dont les tracés ne sont jamais hypsarythmiques : lissencéphalie, syndrome d’Aicardi…
En pratique, les principales situations cliniques peuvent être regroupées en 4 rubriques.
Les spasmes compliquant une encéphalopathie diffuse
Une lésion prénatale diffuse, telle qu’une malformation, est démontrée ou suspectée devant une maladie mentale préexistante ; ou des signes neurologiques. L’EEG est rarement hypsarythmique. Il montre soit un aspect qui est propre au type de lésion cérébrale malformative : agyrie, hémimégalencéphalie, syndrome d’Aicardi : soit des anomalies paroxystiques focales ou multifocales se généralisant dans le sommeil, en cas de lésions corticales multiples : sclérose tubéreuse de Bourneville, séquelles d’anoxo-ischémie du nouveau-né à terme.
Parfois, il n’y a même aucune activité paroxystique, mais un tracé globalement ralenti. Lorsque les rythmes physiologiques sont identifiables, ils sont asymétriques, spontanés ou après administration intraveineuse de diazépam. Chaque spasme est associé à un complexe EEG lent, diffus et de grande amplitude qui précède une diminution importante de l’amplitude du tracé jusqu’au spasme suivant. Le diagnostic repose sur l’anamnèse et les examens cliniques et neuroradiologiques.
Une notion de souffrance périnatale doit être étayée par la mise en évidence de signes directs ou indirects de lésion cérébrale. Parfois, l’anamnèse est négative et la neuroradiologie ne montre aucune lésion cérébrale : ce sont les formes symptomatiques sans lésion identifiable.
La corticothérapie est rarement utile dans ce cas en particulier lorsque l’enfant a présenté, avant les spasmes, des crises généralisées d’un autre type ou des crises partielles avant l’âge de 3 mois ou lorsqu’il n’y a aucune activité paroxystique intercritique : les spasmes persistent de nombreuses années plus tard. Le vigabatrin, le valproate et les benzodiazépines peuvent rendre service.
La lésion focale
La lésion focale Progressive (tumeur angiome) ou non progressive prénatale précoce (dysplasie corticale focale) ou des derniers mois de grossesse (porencéphalie), périnatale (anoxo-ischémie du prématuré) ou post-natale (méningite encéphalite) avec EEG hypsarythmique.
Ces lésions intéressent la région pariéto-occipitale ou rolandique : elles s’étendent parfois sur la région frontale, mais concernent exclusivement la région frontale. Les spasmes ont une composante asymétrique : déviation de la tête et/ou des yeux, oculoclonies, raidissement d’un membre supérieur ou de la main, fixité du regard avant la salve, comme si l’enfant « méditait ses spasmes ».
D’autres types de crises peuvent précéder les spasmes. Il peut exister des signes neurologiques déficitaires focaux. L’évolution des spasmes est souvent favorable sous corticoïdes et cette épilepsie ne modifie pas l’évolution neurologique et cognitive qui dépend de l’étendue et de la topographie des lésions cérébrales préexistantes.
En revanche, l’extension frontale semble indiquer un risque de persistance des spasmes ou l’apparition d’une épilepsie focale ultérieure. Dans la dysplasie corticale focale, l’évolution est souvent défavorable, avec une épilepsie rebelle, voire la persistance de spasmes. L’identification en est parfois difficile sur l’IRM avant que la myélinisation soit complète.
Bien que ses deux premiers groupes soient lésionnels, leur évolution épileptique est très différente.
Les spasmes infantiles cryptogéniques
Les patients ont un développement initial apparemment normal; ils n’ont aucune cause décelable et anomalie au scanner et à l’imagerie par résonance magnétique (IRM), mais une évolution défavorable. La régression est souvent importante avec perte du contact oculaire, voire comportement autistique. Les spasmes ne sont pas précédés d’autres types de crises; ils sont le plus souvent symétriques, mais l’EEG montre des salves de spasmes non indépendants, sous réserve d’un enregistrement assez long, de 12 heures dans certains cas.
Le tracé intercritique est hypsarythmique, souvent asymétrique, en particulier après administration du diazépam. Ces cas sont deux fois sur trois sensibles à la corticothérapie, avec un risque de rechute transitoire dans la deuxième année, en particulier dans le cas où l’EEG au 15e jour de traitement montre la persistance d’un foyer d’ondes lentes et/ou de pointes.
A long terme, la sévérité de l’évolution peut dépendre de la persistance d’une épilepsie ou de troubles cognitifs :
─ certains enfants continuent à avoir des spasmes ; les anomalies EEG diffuses gardent un aspect hypsarythmique ou deviennent plus synchrones, avec des pointe-ondes lentes, réalisant une forme particulière de syndrome de lennox-gastaut; le développement mental est pratiquement arrêté ;
─ d’autres enfants cessent de faire des spasmes mais développent quelques mois ou quelques années plus tard une épilepsie partielle ou généralisée, et gardent un retard mental plus ou moins sévère.
En cas d’épilepsie partielle, le foyer EEG est dans le territoire où les anomalies prédominaient au moment du syndrome de West. Dans ce groupe, les crises ultérieures sont souvent oculo-cloniques ou partielles ou complexes :
─ d’autres enfin cessent d’avoir des crises et leur EEG se normalise ou garde un foyer d’ondes lentes et/ou des pointes.
Qu’ils demeurent ou non épileptiques, la plupart de ces patients gardent des troubles cognitifs et/ou du comportement, de type autistique ou hyperkinétique. Ces troubles semblent être corrélés à la topographie des zones d’hypodébit décelées par le SPECT : temporales en cas de dysphasie et d’hyperkinésie, pariéto-occipitales en cas de troubles visuo-moteurs, et à la fois frontales et temporo-occipitales en cas de comportement autistique.
Ces constatations doivent être comparées à l’évolution du métabolisme chez l’enfant normal : la région pariéto-occipitale devient mature à l’âge habituel d’apparition du syndrome de West.
Dans les 2 premiers cas, le comportement est transitoirement autistique au décours du syndrome de West et cesse de l’être lorsque le moyen de communication épargnée vient à maturité : visuel dans la 1ère année chez les patients avec dysphasie, linguistique dans la 2-3e année chez les patients avec troubles visuo-moteurs.
|
Table des matières
INTRODUCTION
1- Définition
2- Intérêts
3- Objectifs
I- PREMIERE PARTIE : REVUE DE LA LITTERATURE SUR LE SYNDROME DE WEST
1- Définition de l’épilepsie
2- Classification de l’épilepsie
3- Epidémiologie
4- Facteurs étiologiques
5- Facteurs électrocliniques
II- DEUXIEME PARTIE : TRAVAIL PERSONNEL
1- Patients et méthodes
a- Patients
b- Méthodologie
2- Résultats
III- TROISIEME PARTIE : COMMENTAIRE
CONCLUSON
BIBLIOGRAPHIE
Télécharger le rapport complet