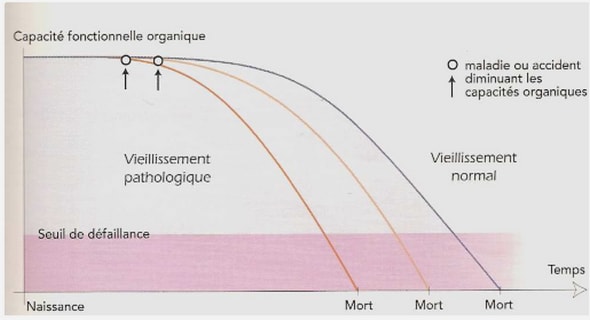
FACTEURS PREDISPOSANTS AU MENINGIOME
Les méningiomes intra-rachidiens
Les méningiomes intra-rachidiens sont des tumeurs généralement bénignes et rares. Ils se développent le plus souvent de manière solitaire à partir d’une prolifération des cellules arachnoïdiennes. Ce sont des tumeurs extra médullaires intra durales ; de siège le plus souvent postérolatéral ou antérolatéral par rapport aux structures radiculo-médullaires. Les méningiomes intra-rachidiens se caractérisent par une croissance lente; ce qui explique l’apparition progressive des signes cliniques. Ces derniers longtemps méconnus peuvent évoluer vers un tableau de compression médullaire, et ils constituent par conséquent une urgence diagnostique et thérapeutique. Devant cet état de fait, le diagnostic du méningiome intra-rachidien doit être évoqué devant tout syndrome de souffrance médullaire ou radiculaire.
L’imagerie par résonnance magnétique (IRM) reste actuellement l’exploration paraclinique de choix du méningiome intra-rachidien, grâce à sa grande performance médullorachidienne. Elle permet d’une part d’assoir le diagnostic en précisant la localisation tumorale et ses rapports avec les structures nerveuses, ainsi qu’avec la dure-mère et d’autre part, elle a également un rôle important dans l’orientation thérapeutique. Les principes du traitement reposent sur l’ablation la plus complète possible du méningiome et de sa base d’insertion dure-mérienne, la difficulté opératoire tient compte surtout de la localisation prémédullaire du méningiome et/ou des formes calcifiées.Les résultats postopératoires sont habituellement satisfaisants avec une franche amélioration fonctionnelle dans 85 %, cependant l’évolution à long terme est marquée par le risque de récidive qui est loin d’être exceptionnelle.
Classification des Méningiomes
Les types histologiques les plus fréquent sont : les méningiomes méningothéliaux et fibreux.La classification de l’OMS sépare les méningiomes en trois grades : le grade 1 est bénin, le grade 2 est atypique alors que le grade 3 est malin. La définition du méningiome atypique se fait par une augmentation de l’activité mitotique ou trois des cinq critères suivants :
Augmentation de la densité cellulaire,
Des foyers de cellules indifférenciées à haut rapport nucléocytoplasmique,
Nucléoles marqués,
Des zones dépourvues de tous dispositif architectural,
Des foyers de nécrose.
Le grade 3, malin se caractérise par des critères histologiques de malignité, c’est-à-dire un nombre de mitoses supérieur ou égal à 20 pour 10 champs a fort grossissement, ou bien un aspect trompeur simulant un sarcome, un carcinome ou un mélanome. L’identification de la variété de méningiome a deux intérêts : diagnostic et pronostique.
Classification des ménigiomes selon l’OMS :
-Méningiome
Méningothélial ;
Transitionnel ;
Fibreux ;
Psammomateux ;
Angiomateux ;
Microkystique ;
Sécrétoire ;
Méningiome à cellules claires ;
Choroide ;
Riche en lymphocytes ;
Métaplasique.
-Méningiome atypique
-Méningiome anaplasique (malin)
GENETIQUE ET BIOLOGIE MOLECULAIRE
C’est principalement grâce au progrès de la biologie moléculaire que les mécanismes de formation des méningiomes sont mieux compris. Généralement des méningiomes sont de tumeurs solitaire, néanmoins, ils peuvent être rencontré dans le cadre d’une maladie autosomique dominante : la neurofibromatose type 2 (NF2), qui comprend plusieurs tumeurs du système nerveux central développées au dépens des enveloppes méningées.Il existe des anomalies des anomalies chromosomiques dans 70 à 75 % des méningiomes. 60 à 70% de ces anomalies sont localisées sur le chromosome 22 et peuvent être de tout type : délétion totale ou partielle du chromosome ou inactivation d’un ou plusieurs gènes.Pour les méningiomes rachidiens l’étude de Markus (40) montre la présence d’anomalies sur le chromosome 22 dans 56% des cas.
Le chromosome 22, est le deuxième plus petit chromosome humain, les études montrent que ce dernier est impliqué dans de nombreuses maladies. En effet, la perte d’information génétique sur le chromosome 22 est observée dans certaines tumeurs comme le phéochromocytome, les différents types de gliomes, les cancers du sein et du colon, les neurinomes et les méningiomes. Ces délétions suggèrent qu’il existe un ou plusieurs gènes suppresseurs tumoraux localisés sur ce chromosome. Les chercheurs sont parvenus à préciser la carte génétique du chromosome 22 et ils ont trouvé que le gène responsable de la neurofibromatose type 2 se situe sur le bras long du chromosome 22.
|
Table des matières
INTRODUCTION
PATIENTS ET METHODES
RESULTATS
I-Données épidémiologiques
1-Fréquence
2-Répartition en fonction de l’âge
3-Répartition en fonction du sexe
4-Répartition en fonction du délai de consultation
II-Données cliniques
1-Symptômes révélateurs
2-Examen clinique
2.1- Syndrome rachidien :
2.2- Syndrome lésionnel:
2.3- Syndrome sous lésionnel :
III-EXAMENS PARACLINIQUES
1-Imagerie
1.1-Imagerie par résonnance magnétique
1.2-Radiographie standard
1.3-Tomodensitométrie rachidienne
2-Bilan biologique
IV-TRAITEMENT
V-ETUDE ANATOMOPATHOLOGIQUE
VI-EVOLUTION
DISCUSSION
I-RAPPELS ANATOMIQUES
II-RAPPELS ANATOMOPATHOLOGIQUES
III-GENETIQUE ET BIOLOGIE MOLECULAIRE
IV-FACTEURS PREDISPOSANTS AU MENINGIOME
V-EPIDEMIOLOGIE
VI-SYMPTOMATOLOGIE CLINIQUE
VII-LOCALISATION DES MENINGIOMES
VIII-EXAMENS PARACLINIQUES
IX-ETUDE HISTOLOGIQUE
X-TRAITEMENT
XI-REEDUCATION
XII-EVOLUTION ET PRONOSTIC
CONCLUSION
ANNEXE
RESUMES
BIBLIOGRAPHIE
![]() Télécharger le rapport complet
Télécharger le rapport complet