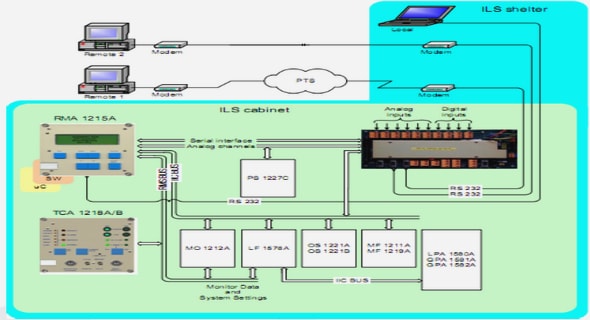
Télécharger le fichier pdf d’un mémoire de fin d’études
Sélection de séquences modèles
Jeux de validation
Extraction des fragments de contacts
Les tables de contingence sont présentées table 2. On constate que si l’on prend une e-value plus stringente à 10 4, on perd de nombreux vrais positifs (441) pour une perte minimale de faux positifs (9). En effet, le rappel passe de 0,74 à 0,50 et même la précision passe de 0,88 à 0,83. Ainsi, ce choix ne semble pas plus approprié. Parmi les faux positifs trouvés avec une e-value à 0,1, il y a 127 séquences eucaryotes dont 44 correspondant à des RdRps eucaryotes, ainsi que diverses enzymes. On retrouve également 58 protéines de virus correspondant essentiellement à des hélicases et protéinases.
HMMER Cette methode a également été utilisée avec un seuil de e-value assez lâche fixé a 0,1. L’aire sous la courbe précision-rappel (figure 7 (b)) est à 0,44. On peut voir comme pour BLAST les 3 mêmes pentes sur la courbe. La première pente descend plus vite que BLAST mais arrive un peu plus tard. En effet, on obtient le premier faux positif pour une e-value de 10 194 alors que l’on a déjà trouvé 70 vrais positifs. La fin de cette première pente se situe autour d’une e-value de 10 98. d’une précision de 0,98 à 0,89. Ces 19 faux positifs sont presque tous des protéinases de virus à ARN simple brin à l’exception de 3 polyprotéines de virus à ARN simple brin contenant des séquences de protéines d’enveloppe et de capside ainsi que des protéases. La deuxième pente arrive pour un rappel légèrement supérieur à 0,40 soit beaucoup plus tôt que pour BLAST. Et on retrouve le même artéfact qu’avec blast. Les tables de contingence sont présentées table 3. De même que pour BLAST, on constate que si l’on prend une e-value plus stringente à 10 4, on perds 102 vrais positifs pour seulement 7 faux positifs. On passe alors d’un rappel à 0,43 à une valeur de 0,38 pour une même précision à 0,94. Ainsi, ce seuil plus stringent ne semble également pas adapté. Concernant les 51 faux positifs trouvés avec une e-value à 0,1, 20 séquences correspondent à des RdRps eucaryotes, mais on a également plusieurs enzymes ou protéines eucaryotes présentant un site de fixation à l’ARN ou l’ADN.
Protomata Pour cette méthode, il a fallu tester différents paramètres p pour trouver celui adapté au nombre de séquences. Ainsi, nous avons gardé ceux présentant les meilleures aires sous la courbe précision-rappel (voir figure A4 et A5 en annexe). Ainsi pour le jeu de paramètre 1.2.1, nous avons choisi p = 10 ayant une aire sous la courbe précision-rappel à 0,65 (figure 7 (c)). Tandis que pour le jeu 8.1.4, nous avons choisi p = 5 qui présente une aire sous la courbe à 0,68 (figure 7 (d)). Pour les deux courbes précision-rappel, on retrouve 2 pentes : la première ressemblant à celle trouvée chez BLAST et HMMER correspond à l’identification de 14 faux positifs ayant de très bons scores et correspondant tous à des protéinases de virus à ARN simple brins. Les deux modèles trouvent les même faux positifs lors de cette pente. La deuxième pente correspond à une augmentation des faux positifs. Elle commence plus tôt pour le jeu 1.2.1 (à savoir pour un plus faible rappel à 0,45 environ contre 0,6 pour le jeu 8.1.4). Il n’y a pas d’artéfact sur ces courbes puisque Protomata a calculé un score pour chaque séquence. Les courbes précision-rappel nous permettent ici de fixer un seuil de score plus adapté que ceux utilisés pour la validation croisée. En effet, il faut trouver les seuils de scores qui optimisent aussi bien le rappel que la précision et pour cela, on peut choisir de se placer sur la courbe juste avant la deuxième pente et trouver les seuils de scores associés à ces valeurs. Ainsi, pour jeu de paramètre 1.2.1, on a choisi un seuil à 21 permettant d’obtenir un rappel à 0.53 et une précision à 0,87. Et pour le jeu 8.1.4, on a choisi un seuil à 35 permettant d’obtenir un rappel à 0.58 et une précision à 0,93. Les tableaux de contingence associés sont présentés table 4.
Dans les 2 cas, les faux positifs trouvés sont essentiellement des protéinases et hélicases de virus à ARN, des RdRps eucaryotes, des enzymes eucaryotes présentant un site de fixation à l’ARN ou l’ADN (par exemple la Ribonuclease J). Le modèle 1.2.1 a également trouvé une reverse transcriptase de retrovirus (protéine P11283).
Discussion En comparant les valeurs statistiques obtenues avec les différentes méthodes, on constate que BLAST présente de meilleurs résultats, il trouve en effet beaucoup plus de vrais positifs sans augmenter énormément le nombre de faux positifs. Ces résultats sont surprenants puisqu’il était attendu que HMMER et Protomata soient plus appropriés pour les séquences vi-rales qui présentent un fort taux de mutation. Concernant HMMER, il est possible au vu de sa courbe précision-rappel 7(b), que la e-value soit plus stringente que BLAST. Il faudrait envi-sager de regarder des seuils encore plus lâches que 0,1. Concernant Protomata, malgré tout le travail de paramétrisation effectué, cet outil ne parvient pas à faire mieux que BLAST sur les polymérases. Ainsi, cet outil n’est peut être pas adapté à l’étude des séquences virales diver-gentes. Concernant les faux positifs, les 3 méthodes retrouvent le même type de séquences : des séquences de virus à ARN non RdRps, des RdRps eucaryotes et d’autres séquences eucaryotes. Ces résultats sont à la fois surprenants et attendus. En effet, dans la littérature les RdRps virales et eucaryotes sont décrites comme partageant peu d’homologie de séquences [19] [8]. Il est donc surprenant de les retrouver aussi facilement. Toutefois, le motif DxDGD, essentiel pour l’activité des RdRps cellulaires, semble être un reste du motif GDD des RdRps virales [19]. Cela met donc en avant une certaine conservation entre les RdRps eucaryotes et celles virales, même si elles appartiennent à des super-familles différentes et cela confirme la difficulté à les distinguer. Egalement, on trouve selon les méthodes, des séquences plus ou moins nombreuses d’enzymes eucaryotes présentant un site de fixation à l’ADN ou l’ARN. Ainsi peut être que la similarité de séquence se situe essentiellement aux niveaux des domaines de fixation à l’ARN ou l’ADN. A l’inverse, un résultat prévisible est le fait de trouver des séquences de virus à ARN n’étant pas des RdRps. Ceci est très certainement dû au fait de travailler sur des poly-protéines comme expliqué dans la partie sur l’étude des séquences modèles de RdRps. Ainsi, même Protomata n’est pas capable de passer cette barrière et pour améliorer les résultats il serait nécessaire de cliver les séquences du jeu modèle pour ne garder que la partie correspondant aux RdRps. Cela permettrait assurément de diminuer le nombre de faux positifs en éliminant toutes les protéinases, hélicases et autres protéines virales identifiées. Et cela permettrait ainsi de faire disparaître la première pente de la courbe précision-rappel. Il est possible que cela influe éga-lement sur la distribution des scores de Protomata et améliore ainsi la discrimination entre les séquences négatives et positives.
|
Table des matières
1 Introduction
2 Matériel et méthodes
2.1 Matériel et méthodes pour les RdRps
2.1.1 Méthodes pour caractériser les RdRps
2.1.2 Sélection de séquences modèles.
2.1.3 Jeux de validation
2.2 Matériel et méthodes pour les protéines de capside
2.2.1 Sélection des structures de capsides
2.2.2 Extraction des fragments de contacts
2.2.3 Adaptation de Yakusa
2.3 Données métagénomiques de Tara Oceans
3 Résultats et Discussion
3.1 Caractérisation et recherche des RdRps
3.1.1 Etude des séquences modèles de RdRps
3.1.2 Sélection des paramètres optimaux pour Protomata par validation croisée
3.1.3 Présentation des résultats – comparaison BLAST – Protomata – HMMER sur jeu de validation
3.2 Caractérisation et étude de la spécificité des capsides de virus
3.2.1 Etude des CFs
3.2.2 Etude de la spécificité des CFs viraux avec Yakusa modifié
3.3 Etude des séquences des données métagénomiques de TARA-Oceans
4 Conclusions
Télécharger le rapport complet