Télécharger le fichier pdf d’un mémoire de fin d’études
Origine géographique et épidémiologie
L’origine du virus est difficile à définir étant donné le manque d’échantillons de patients infectés datés du milieu du XXe siècle. En effet, le cas le plus ancien remonte à 1959, recensé à Kinshasa (anciennement Léopoldville) dans l’actuelle République démocratique du Congo. D’autres cas antérieurs aux années 1980 sont relevés en Europe et aux Etats-Unis.
Cependant, si l’on se réfère à la diversité génétique des souches virales, nous pouvons estimer que plus on observe de variabilité, plus les souches ont eu le temps d’évoluer. Ce raisonnement conduit alors à l’hypothèse d’une origine en Afrique Centrale, compatible avec le fait que selon l’arbre phylogénétique, les souches de VIH-1 M, N et O correspondraient à des transmissions indépendantes du chimpanzé (SIVCPZ) à l’homme. La souche VIH-2 semble en revanche être l’origine de transmissions indépendantes du singe Sooty Mangabey (SIVSM) à l’homme, renvoyant à l’Afrique de l’Ouest (Courgnaud, 2004).
Depuis les années 1990, l’innfection par le VIH-1 est devenue pandémique et est aujourd’hui présente sur toute la surface du globe, alors que le VIH-2 est resté très concentré à la régio Ouest de l’Afrique (Montagnier, 1999).
En 2012, l’ONUSIDA estime à e nviron 35,3 millions le nombre de personnes vivant avec le VIH ; entre 1,9 et 2,7 millions de personnes ont été nouvellement innfectées par le vir. De même, 1,6 million [1,4 millioon– 1,9 million] de personnes sont décédées de causes liées au si dans le monde, contre 2,3 millions [2,1 millions– 2,6 millions] en 20005. Enfin, environ 9,7 millions de personnes vivant avec le VIH ont eu accès au traitement antiréétroviral dans les pays à revenu faible ou intermédiaire (ONUSIDA, fiche d’information mondiale 2013).
Le VIH reste répandu dans plusieurs pays sous-développéscomme l’Innde, mais les pays de l’Afrique sub-saharienne payent un plus lourd tribut.
Structure et génomme
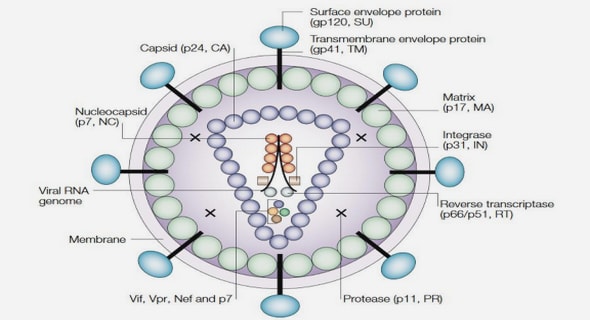
Le VIH est un virus sphérique d’un diamètre moyen d 145 nanommètres, il dispose d’une enveloppe composée d’un fragment de l membrane de la cellule infectée. Dans cette enveloppe lipidique sont ins érés des trimères de glycoprotéine d’ennveloppeEnv). Chaque protéine Env est formée de deux sous-unités : une sous-unité de surfaceg p120 et une sous-unité transmembranaire gp41. Lors de l’attachement du virus à la cellule, la prot éineEnv gp120 se lie à un récepteur CD4présent à la surface des cellules CD4+ du système immuunitaire. À l’intérieur de l’enveloppe, se trouve une maatrice protéique (MA) composée de protéis p17, la capside (CA) composée de protéines p24.Les protéines de la nucléocapside (NC,p7) prrotègent l’ARN viral en le recouvrant. La protéine p6 est exclue de la capside et se trouve entre la matrice et la capside, elle permet la sortie par bouurgeonnement des virus nouvellement formés dans la cellule Le génome du VIH, contenu danns la capside, est constitué d’un simple brin d’ARN’environ 9,6 kb en double exemplaire, acco mpagné d’enzymes : La transcriptase invverse (TI, p66/p51) ou rétrotranscriptase qui rétrotranscrit l’ARN viral en ADN viral. L’intégrrase (IN, p32) qui intèg l’ADN viral à l’ADN cellulaire. La protéase (PR, p12)qui participe à l’assemblage du virus en clivant les précurseurs protéiquesGag p55 et Gag-Pol p160 (Plantier et al., 2009 ; Kuznetsov et al., 2003). Le génome (ARN simple brin) est composé de neuf gènes. Les trois principau sont gag, pol et env, qui définissennt la structure du virus et sont communs à tous les rétrovirus. Les six autres gènes sont tat, rev, neff, vif, vpr et vpu (ou vpx pour le VIH-2), quui codent des protéines régulatrices ayant diverses fonctions(Tableau I).
Immunopathologie de l’infection à VIH
L’immunopathologie du VIH comprend trois phases distinctes: la phase de primo-infection (ou phase aigüe), la phase chronique (ou asymptomatique) et la phase SIDA.
Primo-infection
Après infection de l’organisme, le virus atteint rapidement les nœuds lymphatiques et stimule à la fois la réponse cellulaire et humorale. De plus en plus de cellules sont infectées. Le virus est produit en très grande quantité et entraine en quelques jours une forte chute de globules blancs (leucopénie), et des lymphocytes TCD4+ (Cooper et al., 1985). Deux à quatre semaines après infection, une grande partie du virus est éliminée grâce entre autres aux réponses cellulaires T CD8+ et T CD4+ et des anticorps spécifiques du VIH-1(Figure 5), permettant ainsi une remontée du nombre de lymphocytes T CD4+ (Koup etal., 1994).
Comme observé chez d’autres infections virales, le nombre de cellules T CD8+ augmente lors de la primo-infection (Blumberg and Schooley, 1985). Par ailleurs, une production de cytokines, principalement des cytokines pro inflammatoires et des chimiokines, attirent plus de cellules cibles au site d’infection. Cependant, les cytokines antivirales de la réponse innée (IFNs de Type I) et les cellules T CD8+ de la réponse adaptative arrivent trop tard pour prévenir l’établissement de l’infection (Abel etal., 2005 ; Reynolds et al., 2005). Cette phase est souvent accompagnée de symptômes, non persistants et pouvant passer inaperçus, tels que fièvre, céphalées, myalgies. Le développement desymptômes coïncide avec une virémie plasmatique élevée ayant pour origine une intense réplication du virus dans les organes lymphoïdes
Phase chronique (ou asymptomatique)
La phase dite chronique ou asymptomatique est caractérisée par un contrôle relatif de la réplication virale grâce à la mise en place de fortes réponses spécifiques T CD8 cytotoxiques mais aussi des anticorps spécifiques du VIH-1(figure 5). Cependant la réplication virale persiste à un niveau prédictif de la vitesse de progression de la maladie. Cette phase qui peut durer de 5 à 10 ans, s’accompagne d’une déplétion lymphocytaire T CD4 progressive induite par plusieurs mécanismes:
– la destruction des lymphocytes T CD4 infectés par les cellules T CD8 cytotoxiques spécifiques, l’ADCC et les cellules NK.
– l’apoptose de lymphocytes T CD4 non infectés liée à l’activation chronique
– la pyroptose qui correspond à une forme inflammatoire intense de mort cellulaire
programmée aucours de laquelle le contenu cytoplasmique et les cytokines proinflammatoires sont libérés conduisant à la mort de plusieurs cellules TCD4 (Gilad et al., 2014).
Une activation immunitaire généralisée est notée durant cette phase créant ainsi un environnement propice à la réplication virale. Sans traitement antirétroviral, la destruction progressive des TCD4 conduit au stade SIDA caractérisé par l’apparition d’infections opportunistes et de tumeurs conduisant à la mort du patient.
Phase SIDA
La perte du nombre et de la fonction des cellules TCD4+, résulte d’une très forte diminution de la production de certaines cytokines telles que l’IL-2 responsable de la baisse de l’activité anti-VIH des cellules CD8+. A ce stade, le système immunitaire devient défaillant contre le VIH et les individus VIH+ développent des infections opportunistes ou des tumeurs (Lymphome non-Hodgkinien ou Sarcome de Kaposi) suite à l’altération des défenses immunitaires. Le SIDA est alors déclaré lorsque le nombre de cellules T CD4+ atteint un seuil inférieur ou égal à 200 cellules/mm.
Immunité contre le VIH
L’immunité contre le VIH fait intervenir deux types de réponses : la réponse innée, très rapide et la réponse adaptative pouvant s’établir quelques jours voire des semaines. La réponse innée limite l’action du pathogène, laissant le temps à l’immunité adaptative d’agir de manière beaucoup plus spécifique,
Réponse immunitaire innée
L’immunité innée constitue la première ligne de défense immunitaire et n’est pas spécifique du microorganisme (Janeway et al., 2002). Les cellules impliquées dans la réponse immunitaire innée sont essentiellement les polynucléaires neutrophiles-éosinophiles et basophiles, les monocytes, les macrophages, les cellules dendritiques et les cellules NK « natural killer ». Les épithéliums jouent également un rôle fondamental de première barrière du système immunitaire inné contre les microorganismes. Cette immunité comprend également des médiateurs solubles qui interviennent dans la destruction et l’élimination des microbes.
La reconnaissance des microorganismes par les cellules de l’immunité innée s’effectue via des récepteurs appelésPattern Recognition Receptors (PRRs). Les PRR ont la capacité de reconnaître les motifs microbiens appelés Pathogen-Associated Molecular Patterns (PAMPs). L’activation de la réponse innée induit la production de facteurs extracellulaires. Dans le cas de l’infection par le VIH, les interféronsα et β sont produits en grande quantité par les macrophages et les cellules dendritiques pendant la phase précoce d’infection (Lehner et al., 2011). Ils agissent en potentialisant la réponse immunitaire et peuvent, en particulier pour l’interféronα, empêcher les cellules T d’entrer en apoptose (Cho SS et al., 1996).
D’autres agents solubles de l’immunité innée agissent directement sur le virus, comme les défensines, qui peuvent entre autres inactiver le VIH par un mécanisme de rupture de la membrane virale (Lama and Planelles, 2007). Les molécules du complément et les MBL (Mannose-Binding Lectin) sont aussi capables d’interagir avec le virus et de l’inactiver (Levy, 2011). La réponse innée permet le recrutement de nombreuses cellules immunocompétentes dont les cellules dendritiques plasmacytoïdes qui ont un effet anti-viral par la production d’interférons de type I et de MIP-1α et amplifient les réponses immunes innées et adaptatives par la production d’IFN ɣ, IL15, IL18. Mais ces réponses précoces dans le même temps facilitent la dissémination du pathogène en augmentant localement la quantité de cellules cibles du virus. Comme dans la plupart des infections virales, les cellules NK s’amplifient précocement et participent à la défense immune en lysant des cibles infectées, en sécrétant des cytokines/chimiokines antivirales et en coopérant avec les cellules dendritiques pour la mise en place de réponses T spécifiques car la réponse innée reste insuffisante pour enrayer complètement le VIH.
Réponse immunitaire adaptative
La réponse adaptative repose sur la reconnaissance d’antigènes du pathogène, induisant une destruction directe, par exemple des cellules infectées par des lymphocytes T cytotoxiques CD8+, ou indirecte, comme la production d’anticorps spécifiques du pathogène par des lymphocytes B. Dans ce dernier cas, l’action immunitaire est effectuée soit par activation du complément, soit par liaison de l’anticorps à une cellule immunitaire via des récepteurs aux fragments Fc (FcR). Il semble d’ailleurs que l’infection par le VIH provoque une diminution des réponses cellulaires dépendantes des anticorps, appelées ADCC (Antibody-Dependent Cellular Cytotoxicity) et ADCP (Antibody-Dependent Cellular Phagocytosis), par réduction de l’expression membranaire des FcR (Dugast et al., 2011). Les anticorps dirigés contre le VIH, dits neutralisants, peuvent agir en inhibant la liaison de la protéine d’enveloppe aux récepteurs de la cellule hôte, ou plus tard dans le processus d’entrée du virus, en bloquant l’étape de fusion membranaire (Burton et al., 2004). L’anticorps b12 reconnaît le site de liaison du récepteur CD4 sur la gp120 et constitue le premier compétiteur de la protéine d’enveloppe virale dans l’ordre des étapes du mécanisme d’entrée du virus. La liaison de CD4 engendrant des modifications conformationnelles de la gp120, de nouvelles régions sont exposées et sont reconnues par des anticorps appelés « CD4-induits », comme l’anticorps 17b qui a aidé à la cristallisation de la gp120 (Kwong et al., 1998).
Les cellules T CD4+ effectrices répondent à une infection au VIH-1 par la production de cytokines. Elles sont différenciées en Th1 et Th2, dépendant des cytokines produites. La réponse de type 1 est associée à une forte réponse à médiation cellulaire par la production d’IFNγ, tandis que la réponse de type 2 conduit a une réponse humorale par production d’IL-4 ou IL-10.
Les cellules T CD8+ cytotoxiques réagissent à une variété de peptides viraux Gag( et Nef) (Offenbach et al., 1989). La reconnaissance des peptides Env est faible. Ces cellules T CD8 jouent un rôle majeur en permettant une réduction considérable de la réplication virale pendant la primo-infection en détruisant les cellules dans lesquelles le virus se réplique. Il est clair que ces cellules ont un effet cytopathogène important concourant à la déplétion des lymphocytes T CD4 infectés et à la désorganisation du tissu lymphoïde.
D’autres fonctions protectrices sont assurées par ces cellules par la production de chimiokines (RANTES, MIP- 1α, MIP-1β) qui interagissent avec les co-récepteurs du virus CCR5 et CXCR4. Cependant, le VIH-1 peut échapper aux cellules T cytotoxiques en changeant les epitopes reconnus par celles-ci. Le VIH-1 peut faire ces changements en sélectionnant les virus ayant subit des mutations dans l’épitope reconnu (Ammaranond etal., 2005 ; Price et al., 1997), en diminuant l’expression du CMH (Howcroft et al., 1993) ou en induisant l’anergie des cellules T CD8+ (Klenerman et al., 1996 ; Meier et al., 1995).
En effet, en l’absence de réponses CD4 adéquates lacourse-poursuite entre le virus et les cellules CD8 anti-VIH épuise le système immunitaire par l’activation qu’elle induit. Ceci conduit à l’accumulation de cellules CD8 anti-VIH mémoires très différenciées et activées, ayant perdu l’expression des molécules de co-activation et exprimant de façon très augmentée la molécule inhibitrice PD-1(Programmed cell death protein 1). Ces cellules T CD8 non fonctionnelles pourraient participer à l’incapacité du système immunitaire à contrôler le virus efficacement. E phase terminale la destruction complète de la réponse auxiliaire anti-VIH aboutit à la disparition concomitante de l’ensemble des réponses immunes anti-VIH.
L’infection par le VIH sollicite donc tous les acteurs essentiels du système immunitaire mais la destruction des lymphocytes T CD4, la rapidité d’évolution du virus et l’activation généralisée du système immunitaire qui s’ensuit conduisent la réponse immune spécifique anti-VIH à l’échec.
Traitement et vaccination
Thérapie antirétrovirale
Les traitements antirétroviraux ciblent différentes étapes du cycle du VIH. La zidovudine ou AZT est la première molécule anti-VIH approuvée par les autorités sanitaires en 1987. Durant la dernière décennie, plusieurs molécules ciblant différentes fonctions du VIH ont été évaluées et approuvées pour le traitement du SIDA ; elles sont réparties en six classes (Harris and Bolus, 2008) :
– Inhibiteurs nucléosidiques de la transcriptase inverse (INTI)
– Inhibiteurs non-nucléosidiques de la transcriptase inverse (INNTI)
– Inhibiteurs de la protéase,
– Inhibiteurs de l’intégrase ,
– Inhibiteurs de la fusion,
– Inhibiteurs de l’entrée virale.
En 1996 , la thérapie HAART ( Highly Active Anti-Retroviral Therapy) a été introduite et est le traitement prescrit de nos jours, incluant trois drogues. Des résistances ont été constatées avec la prise du HAART, par conséquent, le traitement doit généralement être suivi et modifié à plusieurs reprises. Ces traitements ne permettent pas d’éradiquer le virus, mais de maintenir en vie les personnes infectées (Stebbing etal., 2006).
Développement d’un vaccin
Aucun vaccin prophylactique efficace n’est disponible contre le VIH. Le développement du vaccin progresse lentement à cause de la variation des souches, ainsi que de la capacité du virus d’échapper à la réponse immunitaire (Goulder et al., 1999). Plusieurs concepts de vaccins se poursuivent, mais les essais n’ont montré aucune protection contre le virus. Jusqu’à présent, les études menées ont montré que le vaccin augmentait le taux de l’infection par le VIH chez ces individus (Sekaly, 2008). Les recherches se poursuivent et l’espoir demeure.
TRAVAIL PERSONNEL
Hypothèse et stratégie de travail
Le système immunitaire est normalement activé en réponse à des agents infectieux et conduit à une prolifération de cellules immunocompétentes avec un contrôle complet de l’infection. L’infection par le VIH provoque une activation persistante du système immunitaire, mais au lieu de contrôler l’infection, cette activation conduit plutôt à un déficit immunitaire, avec une perte progressive de lymphocytes T CD4 + (Hazenberg MD et al., 2000).
L’activation des cellules T est associé à l’apoptose cellulaire (Gougeon ML and Piacentini M 2009) et peut être atténué avec le traitement antirétroviral, mais pas complètement ou au même degré chez toutes les personnes infectées (Nakanjako D etal., 2011; Robbins GK et al., 2009).
L’activation immunologique persistante se reflète à travers l’augmentation de l’expression de marqueurs d’activation de surface des lymphocytes, la production de cytokines pro- inflammatoires et une hausse des taux de prolifération cellulaire et la mort (Mahalingam M etal., 1993 ; Meyaard L et al., 1992). D’où la nécessité d’évaluer cette activation à travers l’expression des différents marqueurs de surface HLA-DR, CD69, CD38. Ces marqueurs sont des antigènes de surface pouvant être exprimé sur les lymphocytes T, B et NK.
HLA-DR, antigène d’histo-compatibilité de classe II est un marqueur tardif d’activation exprimé à la surface des lymphocytes activés suite à une stimulation. CD69 est une protéine de surface induite sur les lymphocytes du sang périphérique activés suite à une stimulation déclenchée à travers la voie d’activation de la protéine C kinase et de la hausse de la concentration de calcium intracellulaire (Krowka JF et al., 1996) . Les transcrits de CD69 sont détectables 30 à 60 minutes après stimulation TCR/CD3 et 2-3 h plus tard, il est exprimé à la surface cellulaire puis atteint son pic entre 18-24 h (Santis AG et al., 1992 ; Lopez-Cabrera M et al., 1993). Dans le sang périphérique, l’antigène CD38 est exprimé par les lymphocytes T, B, les cellules NK et dans une moindre mesure, par les plaquettes et les globules rouges. Parmi les cellules T, il est détectable à des niveaux élevés sur les thymocytes matures et les lymphocytes T activés et à de faibles niveaux sur les cellules au repos (c’est-à-dire HLA-DR- CD25-CD69-) et les cellules naïves (CD45RA+/ R0-) (Mehta K et al., 1996) .
Notre étude a pour objectif d’évaluer l’activation lymphocytaire des patients VIH-1+ par la mesure dans le sang total, des pourcentages de cellules T (TCD4+ et TCD8+) exprimant les marqueurs d’activation HLA-DR, CD69 et CD38 , et l’influence de cette activation sur la progression de la maladie.
Cadre d’étude
Notre étude s’est déroulée au National AIDS Research Institute(NARI) qui est un institut de référence SIDA situé à Pune en Inde où les patients VIH-1 positifs (sous ARV ou non) viennent pour le suivi de leur état immunitaire et font aussi l’objet de plusieurs recherches dans les différents services du centre. Les témoins ont été recrutés parmi le personnel de l’unité d’immunologie dont la séronégativité a été testée. Tous les échantillons ont été manipulés au service d’immunologie et au service de virologie (pour la charge virale).
Matériels et méthodes
Matériels
Matériel de laboratoire
– Tubes avec EDTA ……………………..…………….(Becton Dickinson S.A, Vacutainer)
– Tubes coniques Falcon 15ml, 50ml……..………..………………(Becton Dickinson S.A.)
– Tube Falcon 50ml………………………………….……………………. (BD Biosciences)
– Pipettes sérologiques Falcon 5ml……………………………..…..( Becton Dickinson S.A.)
– Embouts stériles sans filtre 200µl et 1000µl
– Micropipettes (20 µl ,100 µl, 1000 µl)
– Hotte à flux laminaire vertical ……………… ……………….(Envair Electodyne ® , India)
– Centrifugeuse…………………………………………………………(eppendorf ® 5810R )
– Vortex …………………………………………………………………………..(CM101 ® )
– Réfrigérateur à -4°C
– BD FACS Calibur……………………………………………………….. (N o Série E6248)
Réactifs
– Solution de lyse des hématies (1X) (FACS Lysing solution)
– Liquide de gainage (FACS Flow sheath fluid)
– Solution de lavage (FACS Clean)
– Solution de fixation (3.6% Formaldehyde)
– Eau distillée (Sigma water)
– PBS (1X)
– Formaldehyde
– Anti-human CD4 FITC-conjugated
– Anti-human CD4 PerCP-conjugated
– Anti-human CD8 PE-conjugated
– Anti-human HLA-DR PerCP-conjugated
– Anti-human CD69 APC-conjugated
– Anti-human CD38 FITC-conjugated
– Anti-human PD-1(CD279) APC-conjugated
NB: Tous les anticorps monoclonaux couplés aux fluorochromes sont de marque Becton Dickinson S.A.
Population d’étude et matériel biologique
La population d’étude est constituée des patients VIH-1 positifs venant pour un suivi de leur sérologie au National AIDS Research Institute de Pune en Inde où plusieurs recherches sont effectuées. Pour notre étude, nous avons utilisé du sang total prélevé dans un tube EDTA chez 25 individus VIH-1 positifs (6 sous ART et 19 patients LTNP) et 8 témoins négatifs parmi le personnel du service d’immunologie.
Les patients VIH-1 positifs font parti d’une cohorte de recherche au NARI et ont de ce fait signé un consentement libre et éclairé.
|
Table des matières
INTRODUCTION
I- GENERALITES SUR LE VIH
I-1- Historique de découverte du VIH
I-2- Diversité génétique
I-3- Origine géographique et épidémiologie
I-4- Structure et génome du VIH
I-5- Cycle de réplication du virus
I-6- Immunopathologie de l’infection à VIH
I-6-1- Primo-infection
I-6-2- Phase chronique (ou asymptomatique)
I-6-3- Phase SIDA
I-7- Immunité contre le VIH
I-7-1-Réponse immunitaire innée
I-7-2-Réponse immunitaire adaptative
I-8- Traitement et vaccination
I-8-1- Thérapie antirétrovirale
I-8-2- Développement d’un vaccin
II- TRAVAIL PERSONNEL
II-1- Hypothèse et stratégie de travail
II-2- Cadre d’étude .
II-3- Matériel et méthodes
II-3-1- Matériels
II-3-1-1- Matériel de laboratoire
II-3-1-2- Réactifs
II-3-1-3- Population d’étude et matériel biologique
II-3-2- Méthodologie
II-3-2-1- Marquage cellulaire
II-3-2-2- Acquisition et analyse Flow Jo®
II-3-2-3- Analyse statistique des données
II-4- Résultats
II-4-1- Caractéristiques de la population d’étude
II-4-2- Variation des niveaux d’expression des marqueurs
II-4-2-1-Variations des niveaux d’expression des marqueurs chez les témoins négatifs
II-4-2-2- Variations des niveaux d’expression des marqueurs chez les Patients VIH+
II-4-3- Comparaison des proportions de cellules T activées chez les patients VIH+ sous ART versus patients VIH+ LTNP
II-4-4- Comparaison des proportions de cellules T activées entre les témoins négatifs et les patients VIH+
II-4-5- Corrélation entre l’expression des marqueurs d’activation avec le taux de CD4+et la charge virale
II-4-5- 1- Corrélation entre l’expression des marqueurs d’activation et le taux de CD4+
II-4-5-2-Corrélation entre l’expression des marqueurs d’activation et la charge virale
II–5- Discussion
Conclusion et perspectives
Références bibliographiques