Télécharger le fichier pdf d’un mémoire de fin d’études
Le critère principal est un délai
Mon travail de thèse réalisé jusqu’à présent a particulièrement porté sur l’étude de mesures de l’effet traitement lorsque celui-ci est mesuré par la survenue d’un événement au cours du temps. Deux mesures classiques de l’effet traitement et une mesure introduite plus récemment ont été étudiées. Les deux mesures classiques sont la différence de deux estimations de Ka-plan-Meier à un temps donné (DRKM) et le rapport de risques (Hazard ratio, HR) estimé par le modèle de Cox. La nouvelle mesure est une différence de temps de survie moyen borné sans événement (Restricted mean survival time, RMST). Elles sont décrites plus bas.
Différence de risque fournie par deux estimations de Kaplan-Meier (DRKM)
La méthode de Kaplan-Meier permet d’obtenir une courbe de survie et d’estimer les probabi-lités cumulées d’événement à un temps donné. Les tests statistiques du type Logrank permet-tent de comparer des courbes de survie obtenues avec différents traitements. Cette méthode est la plus utilisée lorsqu’aucune hypothèse ne veut être faite sur la distribution des temps de survie. Ici la DR pourrait être estimée par la différence de deux probabilités cumulées d’événement à un temps donné fournies par la courbe de Kaplan-Meier (DRKM) comme pré-senté ci-dessous.
Dans cette courbe de Kaplan-Meier, la probabilité cumulée d’événement à la semaine 96 est de 0.48 dans le groupe 1 et de 0.30 dans le groupe 2. La DRKM est donc de 0.18 (0.48-0.30). Un estimateur de la variance de cette différence permet d’estimer un intervalle de confiance à 95%. Une différence de DRKM avec DR est que la courbe de Kaplan-Meier peut prendre en compte des données censurées, ce qui intervient lorsqu’un patient disparaît d’une étude pour des rai-sons non liées à la conduite de celle-ci ou au traitement reçu (par exemple les patients qui déménagent), c’est-à-dire qu’on ne dispose plus de ses données avant l’apparition de l’événement. On parle de censure « non-informative ». Dans ce cas une hypothèse importante est que la raison du départ des patients de l’étude doit être indépendante de leur risque de l’événement. C’est-à-dire qu’à chaque temps, les patients censurés ont la même perspective de survie que ceux qui continuent d’être suivis. La deuxième notion associée aux données censu-rées est celle de censure non-informative a priori, lorsque les patients sont recrutés sur une longue période de temps et que l’analyse est réalisée à un temps T donné, les participants re-crutés le plus récemment sont censurés lors de l’analyse. C’est une différence importante par rapport à l’analyse de la DR car il ne peut pas y avoir de patients censurés dans l’analyse de la DR, un patient perdu de vue est soit considéré comme un échec soit exclu de l’analyse. En l’absence de censure, l’estimation de Kaplan-Meier au temps t dans un groupe est le nombre de patients ayant eu un événement entre le temps 0 et le temps t divisé par le nombre de pa-tients randomisés dans ce groupe. Dans ce cas, la DRKM au temps t est identique à la DR (P2-P1) au temps t.
La DRKM fournit un résumé interprétable cliniquement des différences entre les groupes à un temps donné et ne nécessite pas d’hypothèse paramétrique pour la distribution des temps de survie dans les deux groupes. Un avantage de la DRKM est qu’elle fournit des informations sur les risques absolus. Cependant, la DRKM ne présente qu’une comparaison à un temps donné sans refléter toute la différence cumulée pour toute la durée d’une étude (Uno et al., 2015).
Le rapport de risques instantanés (Hazard ratio, HR)
Le rapport de risques instantanés est le rapport des risques instantanés dans le groupe recevant le traitement 1 divisé par les risques instantanés dans le groupe recevant le traitement 2. Dans les études cliniques avec un critère de délai jusqu’à un événement, nous étudions la survenue d’un événement binaire en tenant compte de la dynamique temporelle de survenue. Comme décrit plus haut il existe des censures liées à l’interruption du suivi avant la survenue d’événement ou survenant durant la période de suivi, induites par des patients perdus de vue. On utilise le rapport de risques instantanés pour quantifier l’effet du traitement. Par exemple, si le risque instantané de l’événement dans le groupe 1 est de 0.05 et constant dans le temps, et le risque instantané dans le groupe 2 est de 0.025 et également constant dans le temps, le rapport de risques instantanés est 0.025/0.05=0.5. On associe au rapport de risques instantanés un intervalle de confiance. L’interprétation de ce résultat est : le risque instantané de l’événement dans le groupe 2 est seulement la moitié de risque instantané de l’événement dans le groupe 1.
Le rapport de risques instantanés diffère des RR et OR décrits plus haut en ce que les RR et l’OR sont cumulatifs pour toute la durée d’une étude, en utilisant un critère défini, tandis que le rapport de risques instantanés représente les risques instantanés sur une période de l’étude. Dans la pratique, les risques instantanés sont généralement variables en fonction du temps donc le rapport de risques instantanés est une fonction du temps. Le rapport de risques instan-tanés est couramment estimé à l’aide d’un modèle à risques proportionnels de Cox (Case, 2002; Hernán, 2010; Spruance et al., 2004). Ce modèle est une méthode de régression pour les données de survie à la fois puissante et flexible, elle fournit une estimation du rapport de risques et de son intervalle de confiance. Le rapport de risques instantanés qui est dérivé de ce modèle, constitue un résumé valide de la différence entre 2 distributions cumulatives lorsque l’hypothèse des risques proportionnels est respectée. Dans l’hypothèse de risques proportion-nels, l’effet du traitement doit rester constant en fonction du temps. C’est une hypothèse forte du modèle à risques proportionnels de Cox, il faut donc au préalable vérifier que celle-ci est satisfaite. Comme le risque relatif et l’odds ratio, la précision des estimations de rapport de risques instantanés dépend principalement du nombre d’événements observés. (Uno et al., 2015).
Temps de survie moyen borné (Restricted Mean Survival Time, RMST)
La mesure du temps de survie moyen borné a été récemment proposée et consiste à estimer pour un temps T donné, le temps moyen passé sans survenue de l’événement que l’on consi-dère comme critère principal. Techniquement il s’agit d’estimer l’intégrale sous une courbe de Kaplan-Meier comme présenté ci-dessous.
Il est important de spécifier un point temporel de troncature approprié (tau) pour calculer le RMST, qui doit être suffisamment longue pour évaluer ce que nous voulons. Notez que ce tau doit être inférieur au minimum du plus grand temps d’événement observé chez les sujets sui-vis. Lorsque le tau n’est pas spécifié, le minimum du plus grand temps d’événement observé chez les sujets est utilisé par défaut.
Par exemple, dans la figure ci-dessus, nous sommes intéressés à estimer le RMST à la se-maine 96 (tau = 96 semaines). Pour une estimation de Kaplan-Meier faite jusqu’à la semaine 96, l’intégrale sous la courbe peut donner une mesure de temps de suivi moyen sans événe-ment (RMST) de 75 semaines dans le groupe recevant le traitement 1. Ceci indique qu’un futur patient, issu de la même population et recevant le même traitement, suivi également pour une durée de 96 semaines passera en moyenne 75 semaines sans survenue de l’événement. La mesure d’un effet traitement est alors estimée par la différence entre deux moyennes estimées pour deux groupes de traitement. Ici, sachant que le RMST estimé est de 85 semaines dans le groupe 2, cette différence est de 10 semaines en faveur du traitement 2 par rapport au traitement 1, cela indique qu’un patient recevant le traitement 2 passera en moyenne 10 semaines de suivi de plus sans l’événement par rapport à un patient recevant le traitement 1. Un estimateur de la variance de cette différence a été proposé, il permet de tester statistiquement cette différence et d’estimer un intervalle de confiance (Zhao et al., 2012). Par exemple, une différence de RMST entre deux groupes de traitement peut être estimée à 10 semaines avec une variance conduisant à un intervalle de confiance de 7 à 15 semaines.
La mesure RMST a été principalement discutée en oncologie, pour de nombreux types de cancer (Trinquart et al., 2016, 2019a; Uno et al., 2014, 2015). L’analyse basée sur le RMST est considérée comme une mesure récapitulative robuste et interprétable cliniquement qui ne repose pas sur l’hypothèse de risques proportionnels et constitue un événement estimable même sous une forte censure (Royston and Parmar, 2013, 2011; Uno et al., 2015; Zhao et al., 2012). Cependant, la mesure nécessite également une pré-spécification du temps d’intérêt pour permettre aux chercheurs d’envisager la tentation de choisir un temps spécifique afin d’obtenir des résultats plus significatifs.
Les essais de non-infériorité
Depuis plusieurs années, les essais thérapeutiques du VIH ont la particularité d’utiliser quasi-ment exclusivement un design de non-infériorité ou d’équivalence (Flandre, 2015, 2014; Hill and Sabin, 2008). Dans un essai de non-infériorité, l’objectif n’est pas de démontrer la supériorité d’une nouvelle combinaison par rapport à un traitement de référence, mais de montrer que cette nouvelle combinaison n’est pas non-inférieure au traitement standard (Fleming, 2008). Si l’on accepte une certaine perte d’efficacité, ce nouveau traitement doit par ailleurs apporter un avantage par rapport au traitement de référence, par exemple une toxicité moindre, une résistance moindre, ou un coût réduit.
Dans un essai de non-infériorité, les hypothèses nulles et alternatives sont inversées car l’hypothèse nulle est l’infériorité du traitement expérimental par rapport au traitement stan-dard, et l’alternative est la non-infériorité du traitement expérimental par rapport au traitement standard (Blackwelder, 1982; Piaggio et al., 2006). La non-infériorité est démontrée si l’efficacité du traitement expérimental n’est pas trop en deçà de celle du traitement standard. Ce résultat (’en deçà’) se mesure par rapport à une borne de non-infériorité qui correspond donc à la perte maximale d’efficacité acceptée (Fleming, 2008; Piaggio et al., 2006). Le rejet de l’hypothèse nulle (rejet de l’infériorité du traitement expérimental) conduit à conclure à la non-infériorité. Cette non-infériorité s’entend par rapport à une borne ou une limite qui définit la perte maximale d’efficacité admise pour le nouveau traitement par rapport au traitement standard.
La figure ci-dessous présente les scénarios possibles de différence de traitement observée pour des résultats défavorables dans des essais de non-infériorité.
Les bornes de non-infériorité
Comme nous l’avons vu dans la figure ci-dessus, la signification de la borne de non-infériorité est importante : il s’agit de la plus grande perte d’efficacité acceptée par rapport au traitement de référence, par exemple, une borne relative de 10 % signifie que l’on considèrera le nou-veau traitement comme « non-inférieur » tant que son efficacité ne sera pas inférieure de 10 % à celle du traitement de référence. La borne de non-infériorité est définie en fonction du choix de la mesure pour évaluer l’effet du traitement. Dans de nombreux essais thérapeutiques in-cluant des patients infectés par le VIH-1, le critère d’évaluation principal est le plus souvent la proportion de patients en succès (ou en échec) virologique à un temps donné, par exemple à la semaine 48 (“Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents.Department of Health and Human Service.,” n.d.). Dans de tels essais, la DR, mentionnée plus haut, calculée sous la forme d’une simple différence de proportions, est très souvent utilisée comme mesure de l’effet traitement, et les bornes de non-infériorité autour de 10 % à 12 % ont été largement utilisées car cette borne est admise par la Food and Drug administration (Mani et al., 2012). Dans les essais utilisant une analyse basée sur le rapport de risques, les bornes de non-infériorité sont souvent basées sur la DR en raison du nombre important d’essais précédents utilisant la mesure DR. Ces bornes sont ensuite transformées en bornes pour une analyse con-duisant à l’estimation de rapport de risques.
La borne de non-infériorité et de la mesure de l’effet du traitement sont des paramètres essen-tiels pour la conduite et l’analyse des essais thérapeutiques. Ces choix impactent en particulier le calcul de la taille d’échantillon à inclure dans l’essai.
Les essais de l’ACTG
Selon les informations divulguées dans le protocole de l’essai ACTG 5202, la discussion de l’essai a débuté vers 2005/2006 et a publié les résultats en 2011. Par conséquent, la discussion et les recommandations publiées de cette étude sont basées sur le contexte autour de 2011.
La problématique et l’objectif de l’essai
Les recommandations thérapeutiques pour le traitement initial du VIH-1 recommandent 2 inhibiteurs nucléosidiques de la transcriptase inverse(INTI) avec 1 inhibiteur non nucléosi-dique de la transcriptase inverse (INNTI), 1 inhibiteur de protéase (IP) ou 1 inhibiteur d’intégrase (II) (“Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents.Department of Health and Human Service.,” n.d.) (Thompson and Aberg, 2010). Abacavir-lamivudine (ABC/3TC) et tenofovir –emtricitabine (TDF/FTC) sont des traitements INTI efficaces à prise unique quotidienne (DeJesus et al., 2004; Gallant et al., 2006, 2004). Le INNTI le plus utili-sé est efavirenz (EFV), et atazanavir plus ritonavir (ATV/r) est l’un des IP le plus utilisé (“Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents.Department of Health and Human Service.,” n.d.). Il existe peu de comparaisons entre les combinaisons à prise unique quoti-dienne pour le traitement initial du VIH-1. L’essai ACTG (AIDS Clinical Trial Group) 5202 est un essai de non-infériorité dont l’objectif est de comparer l’efficacité et la toxicité de ATV/r versus EFV pour chacune des deux combinaisons ABC/3TC et TDF/FTC, chez les patients naïfs de tout traitement. Un total de 1857 patients éligibles a été inclus dans l’étude entre septembre 2005 et novembre 2007 (Daar et al., 2011). Une comparaison pré-spécifiée de ATV/r et EFV avec les INTIs combinés (plan factoriel) a été effectuée car il n’y avait au-cune preuve que l’effet de ces deux traitements différait en combinant avec les INTIs (p=0.65) (Daar et al., 2011).
Le critère principal de l’essai était le délai jusqu’à un échec virologique. Ce critère était défini comme une charge virale confirmée >=1000 copies/ml à partir de la 16ème semaine et avant la 24ème semaine ou une valeur de charge virale >= 200 copies/ml à partir de la 24ème semaine. L’équipe de l’essai considérait comme acceptable une différence de probabilité d’échec viro-logique d’à peu près 6 % à la semaine 48 et de 10 % à la semaine 96 (cf. supplément (Sax et al., 2009) ). Pour la semaine 96, les hypothèses de taux d’échec virologique étaient de 32 % dans le bras de référence et de 42 % dans un autre bras de traitement. Ceci conduisait à une différence absolue de 10 % (9.69 %) entre les deux bras de traitement. Ces hypothèses con-cernant le taux d’échec virologique étaient basées sur un autre essai ACTG utilisant le zidovu-dine-lamivudine plus l’efavirenz au moment de l’élaboration du protocole (Gulick et al., 2004). Une taille d’échantillon de 450 participants par groupe a été calculée pour fournir une puissance statistique d’environ 90 % permettant de rejeter l’hypothèse nulle d’infériorité, c’est à dire de conclure à la non-infériorité.
Détermination de la borne de non-infériorité
Les bornes de non-infériorité sont définies en premier lieu en termes de différence de risque dans le protocole car elle a été utilisée dans de nombreux essais cliniques antérieurs en raison de sa simplicité (Fischl et al., 2007; Gulick et al., 2006; Riddler et al., 2008).
Le critère principal étant un délai dans l’étude, l’analyse principale utilisait un modèle de Cox pour estimer les différents rapports de risques entre les bras de traitement. Les bornes de non-infériorité devaient donc être transformées en termes de rapport de risques. Pour réaliser cette transformation il faut supposer une distribution pour le temps d’échec virologique, et l’équipe d’ACTG 5202 a supposé une distribution exponentielle. L’hypothèse du modèle exponentiel est celle d’un taux ou d’un risque instantané constant au cours du temps. Si la probabilité cu-mulée de succès est S(t) au temps t, le risque instantané est λ, pour une distribution exponen-tielle nous avons : ( ) = exp(− ) , ∈ (0, ∞) Le risque instantané est donc : = − log( ( )).
Ainsi, si l’on considère une probabilité d’échec de 32 % à la semaine 96, alors la probabilité de succès est de 68 %, et le risque instantané est de 0.004 (-log (0.68)/96). De la même façon pour une probabilité de 42 % le risque instantané est de 0.0056 (-log (0.58)/96). Les bornes d’équivalence pour un rapport de risques sont alors de 0.71 (0.004/0.0056) et 1.40 (0.0056/0.004).
En utilisant les hypothèses faites à la semaine 48 dans le protocole qui correspondent aux taux d’échec de 18 % et 24 %, les risques instantanés sont de 0.004 (-log (0.82)/48) et 0.056 (-log (0.76)/48) respectivement. A noter que l’on obtient les mêmes bornes d’équivalence pour un rapport de risques en utilisant les différentes hypothèses de taux d’échec faites à la semaine 48 et la semaine 96 dans le protocole. En d’autres termes, on constate que les bornes d’équivalence, pour les analyses fondées sur la différence de risque sont distinctes selon une analyse à la semaine 48 ou 96. Au contraire, pour les analyses fondées sur le HR les bornes sont les mêmes quel que soit le temps de l’analyse.
La problématique et l’objectif de l’essai
Les combinaisons antirétroviraux incluant efavirenz (EFV) sont recommandées comme sché-ma thérapeutique de première intention (ou première ligne) par l’Organisation Mondiale de la Santé (OMS) (“WHO | Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection,” n.d.). Cependant, les femmes qui envisagent une grossesse, les patients présentant une résistance préexistante aux traitements inhibiteurs non nucléosidiques de la transcriptase inverse (INNTI) et ceux qui souffrent de troubles psychiatriques graves ne sont pas considérées comme des bons candidats pour un traitement basé sur l’EFV lorsque d’autres options sont disponibles. Pour mieux comprendre l’efficacité et la toxicité des traite-ments alternatifs aux combinaisons d’EFV, l’étude d’ACTG 5257 a été réalisée. Elle compare l’efficacité et la toxicité sur 96 semaines de traitement, chez des patients infectés par le VIH et naïfs de tout traitement, des trois combinaisons suivantes : ténofovir/emtricitabine plus ataza-navir/ritonavir (TDF/FTC/ATV/r), ténofovir/mtricitabine plus raltégravir (TDF/FTC/RAL), et ténofovir/emtricitabine plus darunavir/ritonavir (TDF/FTD/DRV/r) (Lennox et al., 2014). C’est un grand essai de l’équipe ACTG qui suit l’étude ACTG 5202 décrite précédemment dans la partie 3.1.
Le critère principal virologique de l’essai ACTG 5257 était le même que celui défini dans l’essai ACTG 5202, soit une charge virale confirmée >=1000 copies/ml à partir de la 16ème semaine et avant la 24ème semaine ou une valeur de charge virale >= 200 copies/ml à partir de la 24ème semaine. Le critère principal de toxicité était le délai entre la randomisation et l’arrêt de traitement randomisé pour la toxicité. Un critère composite combinant critère virologique et toxicité était aussi analysé.
Détermination de la borne de non-infériorité
L’analyse principale de cet essai était basée sur des comparaisons de la différence entre les estimations de Kaplan-Meier (DRKM) à la semaine 96 avec une équivalence acceptée si l’intervalle de confiance à 97.5 % était entièrement contenu dans la borne -/+10%. L’hypothèse de taux d’échec était de 25% dans le bras de référence à la semaine 96. Cette valeur est motivée par les données de l’étude CASTLE dans laquelle ATV/r plus TDF / FTC présentait un taux d’échec de 22% à la semaine 48 (Molina et al., 2010). Avec cette hypo-thèse de taux d’échec, une taille d’échantillon de 600 participants par groupe a été ciblée pour fournir une puissance statistique d’environ 90 % pour montrer une équivalence dans les com-paraisons par paires. Une approche similaire a été utilisée pour le critère de la toxicité, la mé-thode de Gray (Gray, 1988) était utilisée pour estimer l’incidence cumulative due à la pré-sence de risques compétitifs (Lennox et al., 2014).
Les bornes d’équivalence pour une analyse alternative basée sur le rapport de risques à partir d’un modèle de Cox sont facilement calculées. Comme décrit précédemment dans la partie 3.1, en supposant une distribution exponentielle du délai jusqu’à un échec tel que défini dans le protocole, une probabilité d’échec virologique de 25 % (bras de référence) à la semaine 96 correspond à un risque instantané de 0.003 (-log (1-0.25)/96) et une probabilité de 35 % cor-respond à un risque instantané de 0.0045 (-log (1-0.35)/96). Pour l’échec virologique, la borne d’équivalence supérieure liée à une analyse basée sur le rapport de risques correspondant à la borne de +10 % pour la DRKM est alors de 1.497 (0.0045/ 0.003). De même, la borne d’équivalence inférieure sur la base d’une probabilité d’échec de 15 % est de 0.56 (0.0017/0.003). Les bornes pour le critère de toxicité en supposant une probabilité d’échec de 10 % dans le bras de référence à la semaine 96 est de -/+7 %. De la même façon, les bornes pour une analyse basée sur le rapport de risques sont de 0.29 et 1.77. Dans le protocole ACTG.
Résultats de l’essai
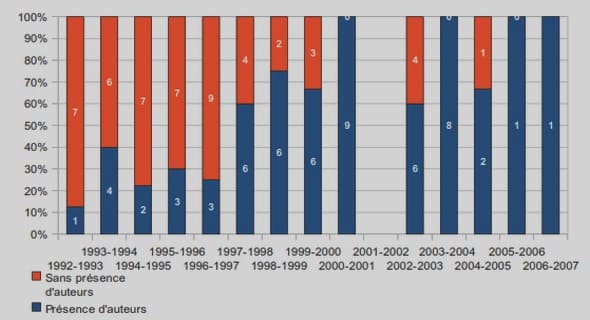
La figure ci-dessous représente les estimations de la DRKM en fonction du critère utilisé.
La probabilité cumulée d’échec virologique estimée par la courbe de Kaplan-Meier à la se-maine 96 était de 12.6 % dans le groupe recevant TDF/FTC/ATV/r, de 9.0 % dans le groupe TDF/FTC/RAL et de 14.9 % dans le groupe recevant le TDF/FTC/DRV/r. Pour toutes les comparaisons de traitement par paires, les intervalles de confiance à 97.5 % se situaient dans la limite d’équivalence prédéfinie de -/+10 %, démontrant l’équivalence des trois régimes par rapport à ce critère d’échec virologique.
|
Table des matières
VALORISATION DES TRAVAUX DE THESE
Article original publié .
Communication affichée
OBJECTIFS
CHAPITRE I – INTRODUCTION GENERALE
CHAPITRE II – METHODES
1. Le critère principal est mesuré à des temps fixes
2. Le critère principal est un délai
CHAPITRE III LES ESSAIS DU VIH
1. Les essais du VIH
2. Les essais de non-infériorité
3. Les essais de l’ACTG
3.1 L’essai ACTG 5202
3.1.1 La problématique et l’objectif de l’essai
3.1.2 Détermination de la borne de non-infériorité
3.1.3 Résultats de l’essai
3.2 L’essai ACTG 5257
3.2.1 La problématique et l’objectif de l’essai
3.2.2 Détermination de la borne de non-infériorité
3.2.3 Résultats de l’essai
CHAPITRE IV CHOIX DE LA MESURE DE L’EFFET TRAITEMENT LORSQUE LES BORNES DE NON-INFERIORITE SONT ORIGINALEMENT DEFINIES PAR UNE DIFFERENCE ABSOLUE TRANFORMEE EN UNE DIFFERENCE RELATIVE A INFLUENCE LES RESULTATS DES ESSAIS CLINIQUES
1. Présentation de l’article
2. Article intégral
2.1 Appendice
2.2 Résultats supplémentaires
CHAPITRE V – L’UTILISATION DU TEMPS DE SURVIE MOYEN BORNE (RESTRICTED MEAN SURVIVAL TIME, RMST) COMME MESURE DE L’EFFET TRAITEMENT DANS LES ESSAIS CLINIQUES SUR LE VIH/SIDA : REANALYSE DE L’ESSAI ACTG5257
1. Présentation de l’article
2. Article intégral
2.1 Appendice
CHAPITRE VI – L’UTILISATION DU TEMPS DE SURVIE MOYEN BORNE (RMST) COMME MESURE DE L’EFFET TRAITEMENT DANS LES ESSAIS SUR LA PROPHYLAXIE PRE-EXPOSITION (PREP)
Manuscrit intégral
CHAPITRE VII – DISCUSSION GENERALE ET PERSPECTIVES
1. Discussion des résultats
2. Enjeux futurs de la recherche
BIBLIOGRAPHIE
Télécharger le rapport complet